 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A structural investigation of the alkali metal site distribution within bioactive glass using neutron diffraction and multinuclear solid state NMR
Richard A.
Martin
*ab,
Helen L.
Twyman
a,
Gregory J.
Rees
c,
Jodie M.
Smith
a,
Emma R.
Barney
d,
Mark E.
Smith
ce,
John V.
Hanna
c and
Robert J.
Newport
a
aSchool of Physical Sciences, Ingram Building, University of Kent, Canterbury, CT2 7NH, UK. E-mail: R.A.Martin@Aston.ac.uk
bSchool of Engineering & Applied Science and Aston Research Centre for Healthy Ageing, University of Aston, Aston Triangle, Birmingham, B4 7ET, UK
cDepartment of Physics, University of Warwick, Coventry, CV4 7AL, UK
dFaculty of Engineering, University of Nottingham, Nottingham, NG7 2RD, UK
eVice-Chancellor's Office, University House, University of Lancaster, Lancaster, LA1 4YW, UK
First published on 26th July 2012
Abstract
The atomic-scale structure of Bioglass and the effect of substituting lithium for sodium within these glasses have been investigated using neutron diffraction and solid state magic angle spinning (MAS) NMR. Applying an effective isomorphic substitution difference function to the neutron diffraction data has enabled the Na–O and Li–O nearest-neighbour correlations to be isolated from the overlapping Ca–O, O–(P)–O and O–(Si)–O correlations. These results reveal that Na and Li behave in a similar manner within the glassy matrix and do not disrupt the short range order of the network former. Residual differences are attributed solely to the variation in ionic radius between the two species. Successful simplification of the 2 < r (Å) < 3 region via the difference method has enabled all the nearest neighbour correlations to be deconvolved. The diffraction data provides the first direct experimental evidence of split Na–O nearest-neighbour correlations in these melt quench bioactive glasses, and an analogous splitting of the Li–O correlations. The observed correlations are attributed to the metal ions bonded either to bridging or to non-bridging oxygen atoms. 23Na triple quantum MAS (3QMAS) NMR data corroborates the split Na–O correlations. The structural sites present will be intimately related to the release properties of the glass system in physiological fluids such as plasma and saliva, and hence to the bioactivity of the material. Detailed structural knowledge is therefore a prerequisite for optimizing material design.
1. Introduction
Melt-quenched silicate glasses containing calcium, phosphorous and alkali metals are of great importance due to their ability to bond chemically to bone and to stimulate new bone growth.1,2 The original, and by far the most commercially successful, bioactive glass is 45S5 Bioglass® developed by Hench and co-workers.1 Bioglass has been in clinical use since 1985 and has already been used in over a million cases.3,4 The composition of Bioglass is (CaO)26.9(Na2O)24.4(SiO2)46.1(P2O5)2.6 (mol%). Under physiological conditions the glass slowly dissolves, releasing calcium and phosphorous into solution which can then redeposit as an amorphous calcium phosphate layer.5The mechanism of bone formation onto a bioactive glass, as described by Hench, is a complex multi-step process.6 The first five steps are given below:
Stage 1. Ion exchange of the alkali and alkali earth elements, typically Na+ and Ca2+, from the glass with H+ from the surrounding physiological fluid:
| Si–O–Na+H2O → Si–OH + Na+(aq) + OH− |
Stage 2. The Si–O–Si are broken to form silanols, releasing soluble Si(OH)4 to the surrounding solution:
| Si–O–Si + H2O → Si–OH + OH–Si |
The first two stages are highly dependent on the overall composition and type of glass (e.g. melt quench vs. sol–gel).
Stage 3. High concentrations of silanol groups (formed during stage 2) are present at the glass![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :solution interface, and begin to re-polymerize to form a silica-rich layer at the glass surface as follows:
:solution interface, and begin to re-polymerize to form a silica-rich layer at the glass surface as follows:

Stage 4. Ca2+ and PO43− groups migrate from the surrounding aqueous medium to the surface, through the silica-rich layer, and precipitate to form a Ca–P2O5 rich film on top of the silica layer. This results in the growth of an amorphous calcium phosphate layer.7
Stage 5. The amorphous calcium phosphate layer begins to incorporate OH− and/or CO32− anions from the surrounding solutions and crystallizes to form a mixed hydroxycarbonate apatite (HCA) layer. The formation of this biologically active HCA layer is believed to be a prerequisite for glasses and glass ceramics to bond to host tissue.
The glass structure, composition and species of ions present strongly influence its chemical durability (i.e. its dissolution in physiological fluids). In turn, the ionic strength and chemical nature of cations present will effect protein–protein interactions.8,9
Each of the components plays a specific role in the dissolution of the glass and the eventual formation of hydroxyapatite. The silica glass includes Ca2+, P5+ and Na+ ions which, via their effect on the glass network structure, control its degradation rate. Calcium and phosphorous dissolve from the glass to create a high concentration solution at the glass:physiological fluid interface, which can then redeposit as an amorphous calcium phosphate – a precursor to the formation of hydroxyapatite. In addition, when P2O5 dissolves it also creates acidic species which help to reduce the pH increase caused by the dissolution of Ca2+ and Na+ ions.10 Furthermore, if suitably controlled via the glass composition, the release rate of Ca, P and Si can stimulate gene transcription in osteoblasts.11 The addition of sodium also lowers the melt temperature and Ca extends the range over which the glass may be worked, helping with glass formation. However, the exact biological role of Na within these glasses is less clear, in fact most bioactive sol–gel derived glasses are sodium free.12–18 Sodium may therefore adopt a primarily structural role within these glasses and be used, in effect, to control the key dissolution rates of Ca, P and Si. However, the precise nature of the local environment surrounding Na ions within these glasses has remained unquantified.
In order to be able to model and predict the behaviour of these materials, and ultimately improve their design, it is necessary to understand the local structure of the glasses. Consequently, the atomic-scale structure of bioactive glasses and its effect on chemical durability and bioactivity has been the focus of much attention.19–23 However the addition of an alkali metal, an alkaline-earth metal as well as a second network former in the form of phosphorous means that the generation of a structural model of Bioglass and its derivatives is challenging. This is particularly true for the sodium environment given that sodium has a low atomic number and its pair correlations are easily masked in a total X-ray diffraction pattern; furthermore, it is mono-isotopic and the advanced structural method of isotopic substitution applied to neutron diffraction, which has been employed to study the local calcium environment,5 is therefore not possible. Although significant progress has been made in understanding the structure of Bioglass by employing molecular dynamic simulations,20–22,24,25 experimental techniques have to date been unable to fully deconvolve the complex overlapping structural features.26
Cations with the same valence and a similar charge-to-size ratio can readily be substituted within the glass matrix; for example, sodium can be replaced with lithium27 or potassium.28,29 It has been suggested that Na–Ca silicate glasses show a non-random distribution of cations, with a preference for Ca–O–Na hetero-bonds at the expense of Ca–O–Ca and Na–O–Na homo-bonds.30 It is therefore important to understand the role alkali metals play in the local environment surrounding calcium ions and determine whether substituting Li for Na affects the Ca–O environment. Recently, by substituting strontium for calcium within these glasses, significant progress has been made on the understanding of the local environment surrounding the calcium ions.31 We have therefore chosen to investigate the influence of substituting Li for Na in the glass series (SiO2)46.1(P2O5)2.6(CaO)26.9(Na2O)24.4−x(Li2O)x (where x = 0, 12.2, 24.4) in terms of the glass' atomic-scale structural characteristics using neutron diffraction and multinuclear 7Li, 23Na, 29Si and 31P solid state magic angle spinning (MAS) NMR.
2. Experimental
2.1. Sample preparation
Melt-quenched glass samples were prepared using SiO2 (Alfa Aesar, 99.5%), P2O5 (Sigma-Aldrich, 98.5%), CaCO3 (Alfa Aesar, 99.95–100.5%) and Na2CO3 (Sigma-Aldrich, 99.5 + %), and/or Li2CO3 (Sigma-Aldrich, 99.99%). The precursors were heated in a platinum crucible to 1400 °C at 10 °C min−1 and held at temperature for 1.5 h. The molten glass was then poured into a pre-heated graphite mould (350 °C) and annealed at this temperature overnight before being allowed to cool slowly to room temperature.7 The batch compositions of the three glasses prepared were (SiO2)46.1(CaO)26.9(P2O5)2.6(Na2O)24.4−x(Li2O)x (where x = 0, 12.2 and 24.4) and they are labelled NaBio, NaLiBio and LiBio respectively. The macroscopic densities of the glass samples were determined by helium pycnometry using a Quantachrome Multipycnometer.2.2. Neutron diffraction method and analysis
Neutron diffraction spectra were collected using the GEM diffractometer at the ISIS spallation neutron source at the Rutherford Appleton Laboratory, UK.32 The coarsely ground samples were held at ambient temperature in a cylindrical vanadium container of 8.3 mm internal diameter and 0.025 mm wall thickness. Interference patterns were collected for each of the samples as well as data for a vanadium rod of 8 mm diameter and the empty GEM instrument in order to perform the appropriate corrections. The data reduction and corrections were performed using the program GUDRUN.32 These corrections involve the removal of background scattering, normalisation, correction for absorption, inelastic and multiple scattering effects and subtraction of the self-scattering term.32 Following these corrections, the resultant coherent scattering intensity, i(Q), is defined by| i(Q) = ∑i ∑jcicjbibj[pij(Q) − 1] | (1) |
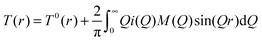 | (2) |
| T0(r) = 4πrρ0(∑icibi)2 | (3) |
Structural information can be obtained by modelling the real-space correlation functions. Pair functions are generated in Q-space and Fourier transformed to allow comparison with the experimental data in real-space. The pair functions are given by:
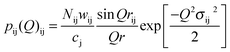 | (4) |
| wij = 2cicjbibj if i ≠ j | (5) |
| wij = ci2bi2 if i = j | (6) |
The method of isomorphic substitution was applied to the neutron diffraction data.33,34 If three total structure factors Nai(Q), NaLii(Q) and Lii(Q) are measured corresponding to scattering lengths bNa > bNaLi > bLi then those correlations not involving the sodium or lithium ion can be eliminated by subtracting two total structure factors to give a first order difference function such as Na–LiΔi(Q) = Nai(Q) − Lii(Q). The addition of the third total structure factor enables three separate first order difference functions to be determined (Na–LiΔi(Q), Na–NaLiΔi(Q) and NaLi–LiΔi(Q)) in order to check the reliability/mutual consistency of the derived data sets. Full details on the method of isomorphic substitution are given by Martin et al.33 The assumption of isomorphism is valid over the short range order, however due to the size difference between the Na+ and Li+ ions the medium and extended range order is not assumed to be isomorphic. The diffraction data is therefore only modelled up to 3 Å.
2.3. Solid state NMR
7Li (I = 3/2) MAS NMR data were acquired at ambient temperatures using a Varian Infinity Plus-300 spectrometer (7.05 T, Larmor frequency of 116.59 MHz) and a Bruker 2.5 mm HX double-air-bearing probe MAS in which spinning frequencies of 27.5 kHz were implemented. A 2 μs π/2 non-selective (or solution) pulse time was calibrated on a primary reference solution of 9.7 M LiCl(aq) (δiso 0.0 ppm), from which a selective (or solids) π/4 pulse time of 1 μs was used in the measurements. A recycle delay of 2 s was used and 16 transients were acquired for each data set. 23Na (I = 3/2) MAS data were acquired using a Bruker AvanceII-600 spectrometer (14.1 T, Larmor frequency of 158.55 MHz). These measurements were undertaken using a Bruker 4 mm HX double-air-bearing probe MAS in which spinning frequencies of 12.5 kHz were implemented. A 5 μs π/2 non-selective (or solution) pulse time was calibrated on a secondary solid reference of NaCl (δiso 7.2 ppm), from which a selective (or solids) π/4 pulse time of 1.25 μs was used in the measurements; recycle delays of 5 s were used. All data are referenced against the primary solution reference of 0.1 M NaCl(aq) (δiso 0 ppm).Corresponding 29Si (I = 1/2) data were acquired on a Varian Infinity Plus 300 system (7.05 T, Larmor frequency of 59.58 MHz) using a Bruker HX 7 mm double-air-bearing probe in which spinning frequencies of 5 kHz were attained. All 29Si spectra were referenced to a secondary solid kaolinite reference which is located at δiso −92 ppm with respect to the primary reference of TMS (δiso 0 ppm, 298 K).35 These data were acquired using single pulse methods with a π/4 pulse time of 2.5 μs and a recycle delay of 30 s. The 31P (I = 1/2) MAS data were measured using a Bruker DSX-400 (9.4 T, Larmor frequency 161.9 MHz) and a Bruker 3.2 mm HX double-air-bearing probe which enabled spinning frequencies of 12.5 kHz. 31P spectra were indirectly referenced to a secondary reference of ammonium dihydrogen phosphate (δiso 0.95 ppm) with 85% H3PO4(aq) being used as the primary reference (δiso 0.0 ppm). These data were also acquired using single pulse methods with a π/4 pulse time of 1.25 μs and a recycle delay of 30 s.31
23Na triple quantum MAS (3QMAS) data were measured on a Bruker AvanceII-600 spectrometer at 158.55 MHz using a double resonance Bruker 4.0 mm HX probe and a MAS frequency of 12.5 kHz. 3QMAS experiments were recorded using a four pulse Z-filter experiment.36,37 A selective excitation pulse (∼3π/2) of 4.5 μs and conversion pulse (∼π/2) of 1.3 μs conversion pulse were followed by two soft non-selective π/2 pulses of 25 μs, and the recycle delay was 5 s.
3. Results and discussion
The glasses were transparent and homogenous with densities of 2.73, 2.70 and 2.66 g cm−3 for NaBio, NaLiBio and LiBio respectively. The increase in density being directly attributed to the increased atomic weight of Na compared to Li.The neutron diffraction interference functions, i(Q), for NaBio, NaLiBio and the LiBio samples are shown in Fig. 1 and the first order difference functions, Δi(Q), are shown in Fig. 2. Fig. 3 shows the corresponding real space pairwise correlation data T(r) obtained by Fourier transforming the i(Q) functions given in Fig. 1, while Fig. 4 shows the real space pairwise correlation data ΔT(r) obtained by Fourier transforming the Δi(Q) functions given in Fig. 2. The weighting factors for each of the total structure factors and difference functions are given in Table 1. The first feature (shortest r-space distance) in T(r), Fig. 3, corresponds to P–O and Si–O correlations (∼1.6 Å), the second feature (∼2.1 to 2.9 Å) contains overlapping O–(P)–O, O–(Si)–O, Ca–O and Na–O and/or Li–O correlations; it is therefore not usually possible to deconvolve these correlations from the real space total diffraction data. However, those correlations that do not contain Na or Li cancel during the formation of the first order difference functions (Table 1). This is confirmed by the notable absence of the P–O and Si–O peaks at ∼1.6 Å in the ΔT(r) functions, illustrated in Fig. 4. Similarly, the O–(P)–O, O–(Si)–O and Ca–O correlations have been eliminated during the formation of the difference functions. Of the remaining correlations present, the first (shortest separation in r-space) correlations will correspond to nearest neighbours Na–O and Li–O. The second nearest neighbours such as Na–(O)–Na, Na–(O)–Li, Na–(O)–P, Na–(O)–Si, Li–(O)–Li, Li–(O)–P and Li–(O)–Si are not expected to occur until beyond 2.8 Å. The first order difference functions may therefore be modelled exclusively using Na–O and Li–O correlations. The data was fitted using the NXFit program developed by Moss.38 It is apparent from Fig. 4 that it is not possible to fit the data using a single Na–O and Li–O correlation, so a two-peak fitting process was employed.
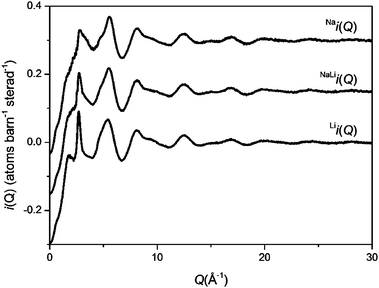 | ||
| Fig. 1 Q-space interference functions, i(Q), for NaBio, NaLiBio and LiBio. The Q-space data extends to 50 Å−1 but only 0 < Q (Å−1) < 30 is shown for clarity. | ||
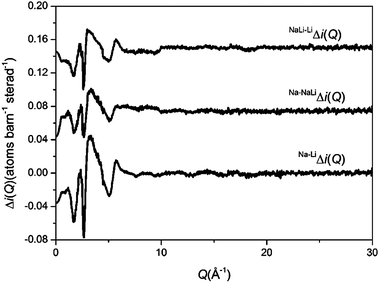 | ||
| Fig. 2 Q-space interference difference functions, Δi(Q), for Na–LiΔi(Q), Na–NaLiΔi(Q) and NaLi–LiΔi(Q). | ||
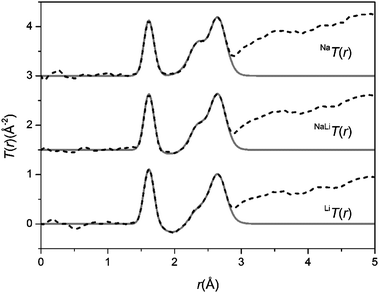 | ||
| Fig. 3 The real-space data for the total diffraction patterns, T(r), the data sets are offset for clarity. The broken curves represent the experimental data and the solid curves are the resultant fits. | ||
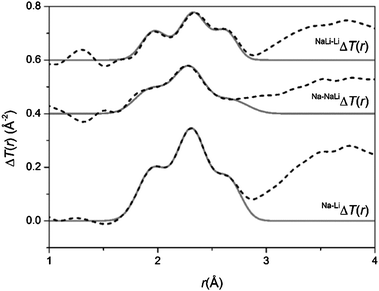 | ||
| Fig. 4 The real-space data for the difference functions, ΔT(r). The broken curves represent the experimental data and the solid curves are the resultant fits. | ||
| Na T(r) | NaLi T(r) | Li T(r) | Na–LiΔT(r) | Na–NaLiΔT(r) | NaLi–LiΔT(r) | |
|---|---|---|---|---|---|---|
| t Na–O(r) | 40.03 | 20.02 | — | 40.03 | 20.02 | 20.02 |
| t Na–Si(r) | 8.43 | 4.22 | — | 8.43 | 4.22 | 4.22 |
| t Na–Ca(r) | 5.57 | 2.79 | — | 5.57 | 2.79 | 2.79 |
| t Na–P(r) | 1.18 | 0.59 | — | 1.18 | 0.59 | 0.59 |
| t Na–Na(r) | 3.90 | 0.98 | — | 3.90 | 2.93 | 0.98 |
| t Na–Li(r) | — | −1.02 | — | 0.00 | 1.02 | −1.02 |
| t Li–O(r) | — | −10.48 | −20.95 | 20.95 | 10.48 | 10.48 |
| t Li–Si(r) | — | −2.21 | −4.41 | 4.41 | 2.21 | 2.21 |
| t Li–Ca(r) | — | −1.46 | −2.92 | 2.92 | 1.46 | 1.46 |
| t Li–P(r) | — | −0.31 | −0.62 | 0.62 | 0.31 | 0.31 |
| t Li–Li(r) | — | 0.27 | 1.07 | −1.07 | −0.27 | −0.80 |
| t Si–O(r) | 43.23 | 43.23 | 43.23 | — | — | — |
| t P–O(r) | 6.03 | 6.03 | 6.03 | — | — | — |
| t O–O(r) | 102.62 | 102.62 | 102.62 | — | — | — |
| t Ca–O(r) | 28.57 | 28.57 | 28.57 | — | — | — |
| t Ca–Si(r) | 6.02 | 6.02 | 6.02 | — | — | — |
| t Ca–P(r) | 0.84 | 0.84 | 0.84 | — | — | — |
| t Ca–Ca(r) | 1.99 | 1.99 | 1.99 | — | — | — |
| t Si–P(r) | 1.27 | 1.27 | 1.27 | — | — | — |
| t Si–Si(r) | 4.55 | 4.55 | 4.55 | — | — | — |
| t P–P(r) | 0.09 | 0.09 | 0.09 | — | — | — |
The output fit parameters for the Na–LiΔi(Q) first order difference function are given in Table 2. The Na–LiΔi(Q) first order difference function has the highest signal:noise ratio, being approximately twice that of the other first order difference functions and therefore provides the most accurate and reliable fit parameters. To ensure the results are self-consistent the other difference functions were modelled using the Na–LiΔi(Q) output parameters as their initial input values for the fitting program and the model was subject to constraints. The resultant output parameters are given in Table 2. This confirms that all the data sets are in agreement and are self-consistent.
| Na–LiΔT(r) | Na–NaLiΔT(r) | NaLi–LiΔT(r) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| r (Å) (±0.02) | N (±0.2) | σ (Å) (±0.04) | r (Å) (±0.02) | N (±0.2) | σ (Å) (±0.04) | r (Å) (±0.02) | N (±0.2) | σ (Å) (±0.04) | |
| Li–O | 1.95 | 3.0 | 0.11 | 1.93 | 3.1 | 0.13 | 1.96 | 2.9 | 0.09 |
| Li–O | 2.27 | 1.6 | 0.21 | 2.25 | 1.7 | 0.17 | 2.29 | 1.5 | 0.24 |
| Na–O | 2.31 | 3.1 | 0.12 | 2.29 | 3.0 | 0.12 | 2.33 | 3.1 | 0.10 |
| Na–O | 2.65 | 1.6 | 0.10 | 2.67 | 1.5 | 0.14 | 2.63 | 1.7 | 0.07 |
Bond valence parameters developed by Brown and Altermatt39 may be used to further verify the Na–O and Li–O parameters obtained from the neutron diffraction fitting. According to the bond valence approach Vi, the oxidation state of cation i, is given by
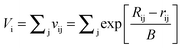 | (7) |
A split Na–O peak has not been directly observed before in bioactive glasses, although this has been predicted in molecular dynamics (MD) simulations for sodium silicates and for Bioglass.40–42 However, MD results for Bioglass are dependent upon the type of simulation employed. For example, classical MD simulations using Shell Models, Rigid Ion Models and Car–Parrinello simulations predicted Na–ONB distances of 2.33, 2.38 and 2.28 Å respectively with between 4.0 and 4.6 oxygen atoms surround each sodium atom, whilst Na–OB distances ranging between 2.45 and 2.52 Å were reported with Na–O coordination numbers between 1.0 and 1.6.41 Na–O coordination numbers reported in MD simulations are higher than presented in this study. However, it is important to note that the majority of MD simulations report Na–O coordination numbers using a cut-off distance of ∼3.1 to 3.3 Å. For example, Tilocca presents Na–O coordination numbers between 5.4 and 5.6 for 45S5 bioglass using a cut-off of 3.1 Å,43 whilst Xiang and Du present a Na–O coordination number of 5.7 using a cut-off of 3.34 Å.20 In the present study the local minima occurs at less than 3 Å (Fig. 4). These correlations are however very broad and the minima does not return to zero, thus by selecting a longer cut-off a larger coordination number would be obtained. It is clear though that the Na–O environment is very disordered and that further correlations appear at distances greater than previously thought. Previous diffraction data reported a Na–O coordination number of six at 2.35 Å,26 it is now clear that the RMC model employed during this study underestimated the O–(Si)–O coordination number, this in turn led to this overestimation of the Na–O coordination number due to the overlapping nature of these correlations. The Na–ONB coordination number of 3.0 presented in this study is however in excellent agreement with previous neutron diffraction and MD results for sodium silicate glasses where values of 3.0 ± 0.5 were reported for the first symmetric peak in T(r).44,45 In crystalline Na2CaSi2O6 a wide range of Na–O distances are reported: 2.30–2.90 Å.46 Similarly a large range of Na–O distances have been reported for MD simulations. Existing simulation models are able to accurately reproduce either the Na–OB or Na–ONB distance but can struggle to reproduce both distances simultaneously. For example, Pedone et al. report Na–ONB and Na–OB distances of 2.32 and 2.42 Å for 45S5 bioglass,47 whilst Malavasi et al. report Na–ONB and Na–OB distances of ∼2.42 and 2.62 Å.42 This study presents experimental distances of ∼2.30 and 2.64 Å for Na–ONB and Na–OB correlations respectively. The Na–O correlations reported here are therefore within the expected values based on existing simulations and experimental data on crystalline analogues. The Li–O values are in similarly good agreement with values reported for crystalline Li-silicates and Na–Li-silicates. Hesse reported 3 oxygen atoms around Li at 1.94 to 1.96 Å with additional oxygen atoms at 2.17 and 2.73 Å for crystalline Li2SiO3.48 Lithium therefore occupies a tetrahedral [LiO4] coordination where one of the Li–O has a very long bond length, the additional oxygen is associated with a neighbouring tetrahedral unit. The results are also in broad agreement with MD simulations e.g. Pedone et al. report a nearest neighbour Li–ONB distance of 1.97 Å and a coordination number of 3.0 in lithium silicates (30 mol% Li2O) and a Li–OB correlation ∼2.09 Å (CN = 0.9).49 The MD simulations therefore report a shorter metal–OB distance for both Na and Li ions.
Fig. 5(a)–(d) shows the solid state 7Li, 23Na, 31P and 29Si MAS NMR, respectively. The 7Li MAS NMR data (see Fig. 5(a)) is represented by single resonances centred at 0.51 ppm and 0.37 ppm for the NaLiBio and LiBio glasses, respectively. The results are in agreement with 7Li NMR studies by Gee et al. where a chemical shift of 0.47 ppm was reported for mixed Na–Li-silicates.50 The introduction of Na to this system shifts the resonances due to next nearest neighbour effects (Na–O–Li), however a minimal effect on the measured nearest neighbour (Li–O) bond lengths is observed (see Table 4). This chemical shift confirms that the alkali metals are mixed in the glass forming Na–O–Li correlations and not phase separated forming solely Li–O–Li correlations.
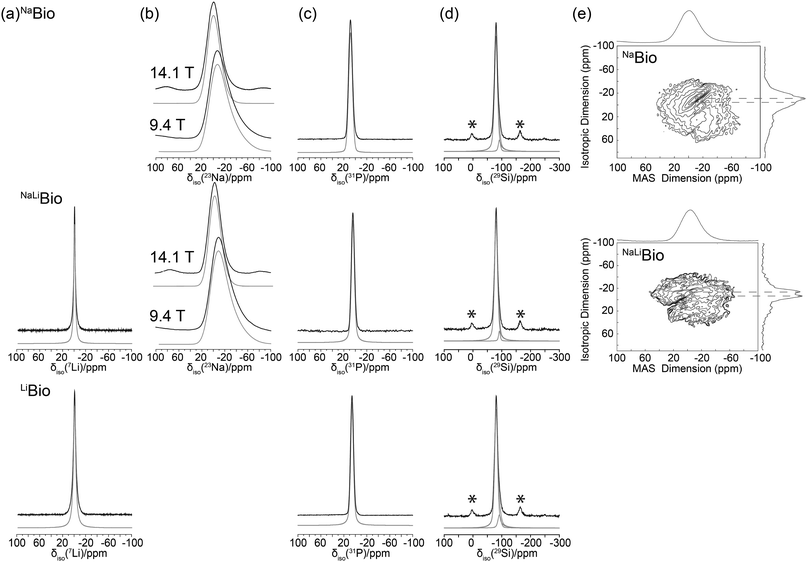 | ||
| Fig. 5 Solid state MAS NMR data for, (a) one pulse 7Li MAS (27.5 kHz) resonances with spectral simulation shown in grey, (b) variable B023Na MAS study with their respected spectral simulations also given in grey, (c) the 31P spectra and its simulations in grey, (d) 29Si study demonstrating the deconvolution into Q2 and Q3 components (* spinning sidebands) and (e) shows the 23Na 3QMAS (12.5 kHz) with their respective projections along the isotropic dimension. | ||
The 23Na MAS NMR data of Fig. 5(b) are represented by single featureless resonances that preclude any detailed insight into the Na speciation that characterises these systems. The simulation of the 23Na data in Fig. 5(b) demonstrates that these species reside in disordered environments, with characteristic tailing of each resonance to high field clearly describing a distribution of field gradients for each glass system. The results of these simulations are summarised in Table 3, with significant differences in the δiso values for these glasses being measured (δiso 7.12 ppm for NaBio and δiso 4.06 ppm for NaLiBio). This upfield shift difference is attributed to an increase in system electronegativity concomitant with Li incorporation. In contrast, the 23Na 3QMAS (see Fig. 5(e)) clearly suggests that more than one Na species is present in each bioglass. The F1 projection spectrum for the NaLiBio system shows three partially resolved resonances in the triple quantum dimension; i.e. one very broad and more diffuse lineshape situated beneath two much narrower resonances. These narrower resonances are not resolved in the F2 dimension. The two narrower resonances for the NaLiBio system may be emerging from differences in the average Na–O bond distances associated with Na–OB and Na–ONB dominated environments, and thus is consistent with the neutron diffraction structural parameters given in Table 2 describing more than one Na environment. Conversely, the 23Na 3QMAS data from the NaBio system shows that much less resolution is afforded in the F1 projection spectrum, although narrower resonances can still be discerned from the broader, more diffuse resonance.
| Bioglass | δ iso/ppm | δ iso/ppm | C Q centre | MHz Width | η Q | δ iso/ppm | Environment/% | δ iso/ppm | Environment/% | δ iso/ppm |
|---|---|---|---|---|---|---|---|---|---|---|
| ±0.05 | ±0.25 | ±0.50 | ±0.50 | ±0.10 | ±0.25 | ±3 | ±0.25 | ±3 | ±0.25 | |
| Nucleus | 7Li | 23Na | 23Na | 23Na | 23Na | 29Si | 29Si | 29Si | 29Si | 31P |
| NaBio | 7.12 | 6.32 | 5.02 | 0.1 | −79.9 | Q2/90 | −94.1 | Q3/10 | 8.0 | |
| NaLiBio | 0.51 | 4.06 | 5.74 | 5.10 | 0.1 | −80.1 | Q2/91 | −93.8 | Q3/9 | 4.1 |
| LiBio | 0.37 | −80.4 | Q2/84 | −94.0 | Q3/16 | 5.6 |
Utilizing the results of both isomorphic substitution and NMR, it is now possible to return to the T(r) (see Fig. 3) and fit the complex overlapping correlations. The short-range order comprises ∼4 oxygens around Si at ∼1.62 Å and ∼4 oxygen around P at a separation of between 1.5 and 1.6 Å, depending on whether the P–O are bridging or non-bridging (also referred to as terminal).51 It is important to note that the first peak in T(r) comprising both the P–O and Si–O correlations is dominated by the Si–O correlation. Due to the low concentration of phosphorous present in these glasses the weighting factor for the Si–O correlation is more than seven times greater than the P–O weighting factor (Table 1). Thus whilst errors associated with the real-space distance, r, are typically ±0.02 Å the error associated with rP–O is much greater. In this case it is therefore not possible to accurately determine the rP–O distance or assign it to bridging or non-bridging P, using diffraction. Instead 31P MAS NMR (discussed below) is used to determine the ratio of bridging and non-bridging P.
The intermediate range order in the range 2.1–2.8 Å consists of overlapping Na–O (and/or Li–O), Ca–O, O–(P)–O and O–(Si)–O correlations and it is therefore not usually possible to resolve these unambiguously. However, by accurately determining the Na–O (and Li–O) correlations using the first order difference method, the remaining region is simplified and requires only Ca–O and O⋯O correlations to be fitted. Furthermore, it is possible to estimate a priori the most probable O⋯O correlations: the O–(P)–O and O–(Si)–O are both tetrahedrally coordinated, therefore the O–(X)–O is given by 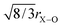 , where X represents P or Si. The concentration of Si is ∼10 times greater than the concentration of P; the O⋯O correlation can therefore be modelled using a single O⋯O correlation that is largely determined by the O–(Si)–O term.
, where X represents P or Si. The concentration of Si is ∼10 times greater than the concentration of P; the O⋯O correlation can therefore be modelled using a single O⋯O correlation that is largely determined by the O–(Si)–O term.
The O⋯O correlation can be calculated using network connectivity models and confirmed using NMR data. For a silicate glass the network connectivity, NC, is given by
| NC = 4 − 2Z, | (8) |
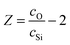 | (9) |
The addition of P complicates the connectivity slightly since P does not behave in an identical manner to Si and eqn (9) cannot therefore be modified simply by substituting cSi+ cP for cSi. For a phosphate-based glass, NC = 3 − y, where y, the number of excess O per P2O5, is given by y = 2cO/cP − 5.52,53 The fact that P is present in relatively small amounts in the glass effectively mitigates the complexities this introduces.
The 31P MAS NMR spectra (Fig. 5(c)) shows an average resonance at δiso ∼ 6(2) ppm representing Q0 species which are essentially isolated PO4 units present throughout the structure. Note, Q1 and Q2 resonances are expected to occur at ∼ −6/−8 and −22/−24 ppm respectively.54 There is no evidence of covalent Si–O–P linkages as suggested by the absence of a significant 31P MAS sideband structure that would invoke the substantial chemical shift anisotropy expected from such environments. This suggests that phosphorous does not enter the network and instead remains in distinct orthophosphate, PO4, units. Although there is an observable shift difference between the NaBio and the NaLi–LiBio. This can be attributed to the previously mentioned increase in electronegativity of Li cation when compared to Na. In this case, since each phosphorous atom is surrounded by 4 non-bridging oxygen atoms, the number of excess oxygen atoms per SiO2 is reduced accordingly and eqn (9) becomes
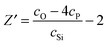 | (10) |
In this case, cO, cSi and cP are 0.552, 0.163 and 0.018 respectively. Therefore, Z′ = 0.944 and the network connectivity of the glass is equal to 2.11. This corresponds to 11% of the Si occupying Q3 connectivity and 89% occupying Q2 connectivity. The experimental 29Si NMR data (see Fig. 5(d)) show asymmetric resonances which can be deconvolved into two Gaussian/Lorentzian (1:1) components located at δ ∼ −80 and −94 ppm; these are assigned to Q2 and Q3 units, respectively. The resultant simulations of these data yield Q3 abundances of 10, 9 and 16% for the NaBio, NaLiBio and the LiBio respectively, with the balance of the Si speciation occupying a Q2 environment. The experimental data is thus in reasonable agreement with the theoretical values provided above (within experimental uncertainties). Knowledge of the Q speciation allows the average O⋯O coordination number to be calculated. A bridging O atom has six next nearest neighbour O atoms whilst a non-bridging O has only three next nearest neighbour O atoms, thus a Si atom having a Q speciation of Q2 or Q3 will have O⋯O coordination numbers of 4 and 4.8 respectively. It can therefore be calculated that the average O–(Si)–O coordination number associated with the samples discussed herein is 4.09. Taking the small concentration of P into account, which is in an orthophosphate environment with a corresponding average O–(P)–O coordination number of 3, the overall average O⋯O coordination number of these glasses is therefore estimated to be 3.98. The O⋯O peak position is expected to occur at ∼2.65 Å, given that 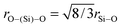 , where rSi–O is 1.62 Å. Thus, the complex region containing overlapping Ca–O, Na–O, O–(P)–O and O–(Si)–O correlations has effectively been further simplified and can now be modelled using the experimentally determined Na–O (or Li–O) input parameters and the O–(P)–O and O–(Si)–O parameters derived above and confirmed using NMR. The only truly unknown feature remaining in this region of the total diffraction pattern (i.e. r < 2.8 Å) is the Ca–O correlation.
, where rSi–O is 1.62 Å. Thus, the complex region containing overlapping Ca–O, Na–O, O–(P)–O and O–(Si)–O correlations has effectively been further simplified and can now be modelled using the experimentally determined Na–O (or Li–O) input parameters and the O–(P)–O and O–(Si)–O parameters derived above and confirmed using NMR. The only truly unknown feature remaining in this region of the total diffraction pattern (i.e. r < 2.8 Å) is the Ca–O correlation.
The fitting parameters for the total diffraction patterns are given in Table 4, and the overall fit is given in Fig. 3 (solid curve). The O⋯O coordination number is consistent with the model outlined above and with recent results given for Ca/Sr bioglass. Hitherto, it has proven difficult to de-convolve diffraction data reliably to estimate the overlapping Na–O, Ca–O and O⋯O correlations; in the absence of additional information, such as that reported here, this resulted in the assumption of a single Na–O correlation and the under-estimation of the O⋯O coordination number.26
| Na T(r) | NaLi T(r) | Li T(r) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| r (Å) (±0.02) | N (±0.2) | σ (Å) (±0.04) | r (Å) (±0.02) | N (±0.2) | σ (Å) (±0.04) | r (Å) (±0.02) | N (±0.2) | σ (Å) (±0.04) | |
| P–O | 1.60 | 4.1 | 0.03 | 1.60 | 3.9 | 0.04 | 1.60 | 3.9 | 0.05 |
| Si–O | 1.62 | 3.9 | 0.07 | 1.62 | 3.9 | 0.07 | 1.62 | 3.9 | 0.07 |
| Li–O | — | — | — | 1.94 | 3.0 | 0.15 | 1.96 | 3.0 | 0.15 |
| Li–O | — | — | — | 2.26 | 1.7 | 0.17 | 2.26 | 1.6 | 0.20 |
| Na–O | 2.30 | 3.0 | 0.16 | 2.32 | 3.0 | 0.16 | — | — | — |
| Na–O | 2.64 | 1.5 | 0.10 | 2.66 | 1.7 | 0.14 | — | — | — |
| Ca–O | 2.36 | 5.3 | 0.10 | 2.36 | 5.3 | 0.11 | 2.36 | 5.3 | 0.13 |
| O–O | 2.65 | 4.1 | 0.11 | 2.65 | 4.0 | 0.11 | 2.65 | 4.1 | 0.11 |
| Ca–O | 2.72 | 1.2 | 0.17 | 2.73 | 1.4 | 0.15 | 2.74 | 1.2 | 0.19 |
The Ca–O values presented in Table 4 are in excellent agreement with recently reported values obtained using an isomorphic substitution of Sr for Ca where Ca–ONB and Ca–OB distances of ∼2.36 Å and 2.72 Å were reported.31 The values are consistent with crystalline Na2CaSi2O6 for which Ca–O values in the range 2.32–2.72 Å are reported.46 Interestingly, the substitution of Na with Li does not appear to significantly influence the local Ca–O environment. This is important since previous results suggested that Na–Ca silicate glasses show a non-random distribution of cations, with a preference for Ca–O–Na bonds at the expense of Ca–O–Ca and Na–O–Na bonds.30 It may therefore also be expected that a preference for Ca–O–Li bonds may exist.
In contrast, in sol–gel analogues containing no Na+ (or Li+) but which do contain significant levels of H+, an additional Ca–O correlation at ∼2.5 Å is found which is assigned to Ca–OH.5,55 Whilst hydroxyl groups are broadly assumed to play a similar structural role in sol–gel derived bioactive glasses to that of Na (or Li) in melt quench glasses (i.e. provide a route for incorporating additional oxygen atoms to break up the silica network connectivity and to provide a small monovalent ion that easily leaches from the glass under aqueous/physiological conditions), it appears –OH has a much more pronounced effect on the local environment surrounding the calcium ions.
Solubility (and therefore bioactivity) of these glasses is controlled by 3 key factors. The average network connectivity determined by the oxygen to silicon ratio (eqn (8) and (9)) is the most important parameter. The second term relates to the valency of the modifying ions such that monovalent systems are more soluble than divalent systems, i.e. a sodium silicate will be more soluble than a calcium silicate with the same network connectivity. Finally the cation size influences the solubility due to the expansion of the glass matrix. Although the network and short range order (e.g. Si–O and O–Si–O) are not significantly affected by the alkali ion substitution the increase in cation size expands the matrix through longer Na–O correlations compared to Li–O correlations. The expanded O–Na–O correlations compared to the O–Li–O correlations will result in a more open structure which will increase the rate of dissolution. Analogous results have been reported when Sr is substituted for Ca within bioactive glasses.56 Note the mechanism for dissolution will not apply to sol–gel derived glasses due to the much wider Qn distribution and silanol groups present.57 Therefore glasses containing H+ would dissolve faster than equivalent Li+ or Na+ glasses.
The key assumption underpinning the isomorphic substitution method employed is that the short range order pair correlations not containing the isomorphs, in this case Na/Li, are not significantly affected by the substitution and that these correlations therefore successfully cancel during the formation of the first order difference function. As shown in Table 3, the correlations not containing Na or Li are consistent for each of the three total diffraction patterns. It is apparent from the fitting parameters returned that the model is fully self-consistent between the experimental values for the first order difference functions, the total diffraction data, the solid state MAS NMR data and theory.
4. Conclusions
The diffraction data presented and discussed above affords a detailed structural model of the local and intermediate range order in bioactive glasses wherein the study of the Na site is made more tractable by substituting Li for Na within these systems. The conclusions drawn from diffraction studies are strongly supported by the complementary NMR data. The assumption that isomorphic substitution could be employed to determine the short range order of Na and Li bioactive glasses is thereby verified. The Na–O environment for the present melt-quench glass was found to be very disordered and was best modelled using two Na–O correlations centred at 2.31 Å and 2.65 Å. This provides the first direct experimental evidence of a split Na–O correlation in Bioglass and supports existing molecular dynamics simulations. The substitution of Na with Li does not appear to affect the Ca–O local environment. Bioglass is a complex glass system, containing 5 separate elements and therefore 15 overlapping pair correlation functions: the present study therefore represents one of the most detailed studies undertaken on any complex glassy system and has resulted in the characterization of the short and intermediate range order.Acknowledgements
This work was supported by EPSRC grants EP/E050611/1 and EP/E051669/1. The authors wish to thank the STFC for the provision of beam-time at the ISIS pulsed neutron source. We wish to acknowledge the use of the Chemical Database Service at Daresbury. RAM acknowledges the Royal Society for the Research Grant RG100147. JVH and MES acknowledge the continued funding of the 300, 400 and 600 MHz instrumentation used in this research which was facilitated by EPSRC, the University of Warwick and partial funding through Birmingham Science City Advanced Materials Projects 1 and 2 supported by Advantage West Midlands (AWM) and the European Regional Development Fund (ERDF).References
- L. L. Hench, R. J. Splinter, W. C. Allen and T. K. Greenlee, J. Biomed. Mater. Res. Symp., 1971, 5, 25 CrossRef.
- A. E. Clark, L. L. Hench and H. A. Paschall, J. Biomed. Mater. Res., 1976, 10, 161 CrossRef CAS.
- L. L. Hench, J. Mater. Sci. Mater. Med., 2006, 17, 967 CrossRef CAS.
- L. L. Hench, W. June and D. C. Greenspan, J. Australas. Ceram. Soc., 2004, 40, 1 CAS.
- L. J. Skipper, F. E. Sowrey, D. M. Pickup, K. O. Drake, M. E. Smith, P. Saravanapavan, L. L. Hench and R. J. Newport, J. Mater. Chem., 2005, 15, 2369 RSC.
- L. L. Hench and I. Thompson, J. R. Soc. Interface, 2010, 7, S379 CrossRef CAS.
- R. A. Martin, H. Twyman, D. Qiu, J. C. Knowles and R. J. Newport, J. Mater. Sci. Mater. Med., 2009, 20, 883 CrossRef CAS.
- F. J. Zhang, M. W. A. Skoda, R. M. J. Jacobs, R. A. Martin, C. M. Martin and F. Schreiber, J. Phys. Chem. B, 2007, 111, 251 CrossRef CAS.
- L. Ianeselli, F. J. Zhang, M. W. A. Skoda, R. M. J. Jacobs, R. A. Martin, S. Callow, S. Prevost and F. Schreiber, J. Phys. Chem. B, 2010, 114, 3776 CrossRef CAS.
- I. Ahmed, M. Lewis, I. Olsen and J. C. Knowles, Biomaterials, 2004, 25, 491 CrossRef CAS.
- I. D. Xynos, A. J. Edgar, L. D. K. Buttery, L. L. Hench and J. M. Polak, J. Biomed. Mater. Res., 2001, 55, 151 CrossRef CAS.
- Y. Minaberry and M. Jobbagy, Chem. Mater., 2011, 23, 2327 CrossRef CAS.
- I. Izquierdo-Barba, D. Arcos, Y. Sakamoto, O. Terasaki, A. López-Noriega and M. A. Vallet-Regí, Chem. Mater., 2008, 20, 3191 CrossRef CAS.
- J. Román, S. Padilla and M. Vallet-Regí, Chem. Mater., 2003, 15, 798 CrossRef.
- D. Arcos, D. C. Greenspan and M. Vallet-Regi, Chem. Mater., 2002, 14, 1515 CrossRef CAS.
- P. Sepulveda, J. R. Jones and L. L. Hench, J. Biomed. Mater. Res., 2002, 59, 340 CrossRef CAS.
- V. FitzGerald, R. A. Martin, J. R. Jones, D. Qiu, K. M. Wetherall, R. M. Moss and R. J. Newport, J. Biomed. Mater. Res., Part A, 2009, 91A, 76 CrossRef CAS.
- R. A. Martin, S. Yue, J. V. Hanna, P. D. Lee, R. J. Newport, M. E. Smith and J. R. Jones, Philos. Trans. R. Soc. London, Ser. A, 2012, 370, 1422 CrossRef CAS.
- A. Pedone, T. Charpentier, G. Malavasi and M. C. Menziani, Chem. Mater., 2010, 22, 5644 CrossRef CAS.
- Y. Xiang and J. Du, Chem. Mater., 2011, 23, 2703 CrossRef CAS.
- A. Tilocca, A. N. Cormack and N. H. de Leeuw, Chem. Mater., 2007, 19, 95 CrossRef CAS.
- R. N. Mead and G. Mountjoy, Chem. Mater., 2006, 18, 3956 CrossRef CAS.
- J. M. Oliveira, R. N. Correia and M. H. Fernandes, Biomaterials, 2002, 23, 371 CrossRef CAS.
- M. Corno, A. Pedone, R. Dovesi and P. Ugliengo, Chem. Mater., 2008, 20, 5610 CrossRef CAS.
- A. Tilocca, J. Mater. Chem., 2010, 20, 6848 RSC.
- V. FitzGerald, D. M. Pickup, D. Greenspan, G. Sarkar, J. J. Fitzgerald, K. M. Wetherall, R. M. Moss, J. R. Jones and R. J. Newport, Adv. Funct. Mater., 2007, 17, 3746 CrossRef CAS.
- E. Kuzielová, M. Palou, J. Lokaj and J. Kozánková, Adv. Appl. Ceram., 2008, 107, 203 CrossRef.
- I. Elgayar, A. E. Aliev, A. R. Boccaccini and R. G. Hill, J. Non-Cryst. Solids, 2005, 351, 173 CrossRef CAS.
- A. Angelopoulou, V. Montouillout, D. Massiot and G. Kordas, J. Non-Cryst. Solids, 2008, 354, 333 CrossRef CAS.
- S. K. Lee and J. F. Stebbins, J. Phys. Chem. B, 2003, 107, 3141 CrossRef CAS.
- R. A. Martin, H. Twyman, G. J. Rees, E. R. Barney, R. M. Moss, J. M. Smith, R. G. Hill, G. Cibin, T. Charpentier, M. E. Smith, J. V. Hanna and R. J. Newport, submitted.
- A. C. Hannon, Nucl. Instrum. Methods Phys. Res., Sect. A, 2005, 551, 88 CrossRef CAS.
- R. A. Martin, P. S. Salmon, H. E. Fischer and G. J. Cuello, J. Phys.: Condens. Matter, 2003, 15, 8235 CrossRef CAS.
- R. A. Martin, P. S. Salmon, H. E. Fischer and G. J. Cuello, Phys. Rev. Lett., 2003, 90, 185501 CrossRef.
- R. K. Harris, E. D. Becker, S. M. C. de Menezes, R. Goodfellow and P. Granger, Solid State Nucl. Magn. Reson., 2002, 22, 458 CrossRef CAS.
- J.-P. Amoureux, C. Fernandez and S. Steuernagel, J. Magn. Reson., Ser. A, 1996, 123, 116 CrossRef CAS.
- M. E. Smith and E. R. H. van Eck, Prog. Nucl. Magn. Reson. Spectrosc., 1999, 34, 159 CrossRef CAS.
- R. M. Moss, PhD thesis, University of Kent, 2009.
- I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 244 CrossRef.
- A. N. Cormack and J. C. Du, J. Non-Cryst. Solids, 2001, 293, 283 CrossRef.
- A. Tilocca, J. Chem. Phys., 2008, 129, 084504 CrossRef.
- G. Malavasi, M. Menziani, A. Pedone, B. Civalleri, M. Corno and P. Ugliengo, Theor. Chem. Acc., 2007, 117, 933 CrossRef CAS.
- A. Tilocca, N. H. de Leeuw and A. N. Cormack, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 104209 CrossRef.
- N. M. Vedishcheva, B. A. Shakhmatkin, M. M. Shultz, B. Vessal, A. C. Wright, B. Bachra, A. G. Clare, A. C. Hannon and R. N. Sinclair, J. Non-Cryst. Solids, 1995, 193, 292 CrossRef.
- B. Vessal, A. C. Wright and A. C. Hannon, J. Non-Cryst. Solids, 1996, 196, 233 CrossRef CAS.
- H. Ohsato, I. Maki and Y. Takeuchi, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1985, 41, 1575 CrossRef.
- A. Pedone, G. Malavasi and M. C. Menziani, J. Phys. Chem. C, 2009, 113, 15723 CAS.
- K. F. Hesse, Acta Crystallogr., Sect. B: Struct. Sci., 1977, 33, 901 CrossRef.
- A. Pedone, G. Malavasi, A. N. Cormack, U. Segre and M. C. Menziani, Chem. Mater., 2007, 19, 3144 CrossRef CAS.
- B. Gee, M. Janssen and H. Eckert, J. Non-Cryst. Solids, 1997, 215, 41 CrossRef CAS.
- U. Hoppe, G. Walter, A. Barz, D. Stachel and A. C. Hannon, J. Phys.: Condens. Matter, 1998, 10, 261 CrossRef CAS.
- R. A. Martin, P. S. Salmon, C. J. Benmore, H. E. Fischer and G. J. Cuello, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 054203 CrossRef.
- R. A. Martin, P. S. Salmon, H. E. Fischer and G. J. Cuello, J. Non-Cryst. Solids, 2004, 345–346, 208 CrossRef.
- D. Carta, D. M. Pickup, J. C. Knowles, I. Ahmed, M. E. Smith and R. J. Newport, J. Non-Cryst. Solids, 2007, 353, 1759 CrossRef CAS.
- R. J. Newport, L. J. Skipper, V. FitzGerald, D. M. Pickup, M. E. Smith and J. R. Jones, J. Non-Cryst. Solids, 2007, 353, 1854 CrossRef CAS.
- Y. C. Fredholm, N. Karpukhina, D. S. Brauer, J. R. Jones, R. V. Law and R. G. Hill, J. R. Soc. Interface, 2012, 9, 880 CrossRef CAS.
- K. Fujikura, N. Karpukhina, T. Kasuga, D. S. Brauer, R. G. Hill and R. V. Law, J. Mater. Chem., 2012, 22, 7395 RSC.
| This journal is © the Owner Societies 2012 |