
A theoretical simulation on the catalytic oxidation of CO on Pt/graphene†
Abstract
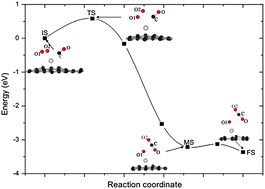
The catalytic oxidation of CO on Pt/X-graphene (X = “pri” for pristine- or “SV” for defective-graphene with a single vacancy) is investigated using the first-principles method based on density functional theory. In contrast to a Pt atom on pristine graphene, a vacancy defect in graphene strongly stabilizes a single Pt adatom and makes the Pt adatom more positively charged, which helps to weaken the CO adsorption and facilitates the O2 adsorption, thus enhancing the activity for CO oxidation and alleviating the CO poisoning of the platinum catalysts. The CO oxidation reaction on Pt/SV-graphene has a low energy barrier (0.58 eV) by the Langmuir–Hinshelwood (LH) reaction (CO + O2 → OOCO → CO2 + Oads) which is followed by the Eley–Rideal (ER) reaction with an energy barrier of 0.59 eV (CO + Oads → CO2). The results validate the reactivity of catalysts on the atomic-scale and initiate a clue for fabricating carbon-based catalysts with low cost and high activity.
- This article is part of the themed collection: Computational Catalysis and Materials for Energy Production, Storage and Utilization
 Please wait while we load your content...
Please wait while we load your content...

