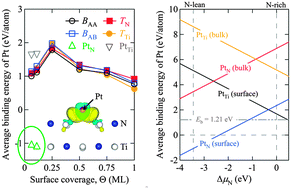
As a first step towards a microscopic understanding of single-Pt atom-dispersed catalysts on non-conventional TiN supports, we present density-functional theory (DFT) calculations to investigate the adsorption properties of Pt atoms on the pristine TiN(100) surface, as well as the dominant influence of surface defects on the thermodynamic stability of platinized TiN. Optimized atomic geometries, energetics, and analysis of the electronic structure of the Pt/TiN system are reported for various surface coverages of Pt. We find that atomic Pt does not bind preferably to the clean TiN surface, but under typical PEM fuel cell operating conditions, i.e. strongly oxidizing conditions, TiN surface vacancies play a crucial role in anchoring the Pt atom for its catalytic function. Whilst considering the energetic stability of the Pt/TiN structures under varying N conditions, embedding Pt at the surface N-vacancy site is found to be the most favorable under N-lean conditions. Thus, the system of embedding Pt at the surface N-vacancy sites on TiN(100) surfaces could be promising catalysts for PEM fuel cells.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


