 Open Access Article
Open Access Article
Received
30th June 2011
, Accepted 4th October 2011
First published on 28th October 2011
Abstract
Time-resolved vibrational spectroscopy is used to investigate the inter-component motion of an ultraviolet-triggered two-station molecular shuttle. The operation cycle of this molecular shuttle involves several intermediate species, which are observable in the amide I and amide II regions of the mid-IR spectrum. Using ab initio calculations on specific parts of the rotaxane, and by comparing the transient spectra of the normal rotaxane with that of the N-deuterated version, we can assign the observed vibrational modes of each species occurring during the shuttling cycle in an unambiguous way. The complete time- and frequency-dependent data set is analyzed using singular value decomposition (SVD). Using a kinetic model to describe the time-dependent concentrations of the transient species, we derive the absorption spectra associated with each stage in the operation cycle of the molecular shuttle, including the recombination of the charged species.
1 Introduction
Recent developments in supramolecular chemistry have led to the synthesis of a multitude of molecular devices capable of mimicking the function of macroscopic machinery. They include molecular shuttles,2–4 propellers,5 gears,6 ratchets,7 valves,8 turnstiles,9 motors,10–13 brakes,14 and elevators.15 Inducing and controlling the motion of such devices is of fundamental interest if these devices are to be used as a basis for the construction of more complex and useful molecular machines. However, the analogy between molecular devices and their macroscopic counterparts, appealing as it may be, does not hold at a molecular level. Macroscopic concepts such as viscous friction lose meaning as the size and speed of motion of the device approach that of solvent molecules.16–19 Also, random thermal motion, both of the surrounding solvent molecules and of the components of the molecular machines themselves, will inevitably lead to a certain degree of unpredictability in the device's motion.
Observing the underlying elementary events of the motion of molecular machines requires a technique with high structural sensitivity as well as sufficient time resolution. Most spectroscopic methods that have been used previously to investigate molecular machines (X-ray reflectometry,20 circular dichroism,11 steady-state absorption spectroscopy,21–25cyclic voltammetry,8,15,21 and NMR26) do not provide sufficient time resolution to observe the devices in operation. Transient UV-VIS spectroscopy2 has the required time resolution, but probes the conformation in an indirect manner. Time-resolved vibrational spectroscopy is an experimental method which provides both structural sensitivity and temporal resolution.27–30 In particular, the possibility to probe the stretching modes of specific, localized, chemical bonds allows for a direct view on conformational changes at the molecular level, making it possible to probe the individual components of a molecular machine and their interactions in a direct manner. The time resolution of the probing is determined by the duration of the IR-probe pulse, and can in principle be as short as 100 fs.31
We have demonstrated recently how time-resolved UV-pump IR-probe experiments can be used to unravel the operation mechanism of rotaxane-based molecular shuttles.32 Here, we report more extensive UV-IR experiments, in which we investigate a larger range of both IR-probe frequencies and time delays. We confirm our assignments using ab initio calculations on (parts of) the rotaxane in each stage of the operation cycle. We also perform a detailed analysis of the kinetics using singular value decomposition (SVD). In this way, we determine the number of significant spectral and temporal components in the data in an objective manner.33,34 From the SVD, we reconstruct the vibrational spectrum of the rotaxane shuttle in each stage of its operation cycle.
Rotaxanes are compounds that consist of a macrocycle that is mechanically interlocked onto a linear thread.35 The structure of the rotaxane-based molecular shuttle studied here is shown in Fig. 1. There are two specific positions (stations) on the thread at which the macrocycle (blue in Fig. 1) can form hydrogen bonds. These hydrogen bonds are formed between the NH groups of the macrocycle and the CO groups of the thread. The naphthalimide station (ni, grey in Fig. 1) is a poor hydrogen-bond acceptor in its electronic ground state.36 Therefore, in the neutral rotaxane, the ring resides predominantly (>99%) on the succinamide station (succ, green in Fig. 1). The macrocycle can be induced to move from the succ to the ni station by means of an electrochemical or photochemical reduction of the ni station. In the latter case, excitation of the naphthalimide station with a 355 nm pulse results in rapid (τ = 1.6 ns) intersystem crossing to the triplet state.37 In this state, the ni station can be reduced by an external electron donor to form a radical anion. In the radical anion state, the naphthalimide station (ni˙−, red in Fig. 1) has a much higher hydrogen-bonding affinity towards the macrocycle than the succ station (equilibrium constant >1000).3 As a consequence, the macrocycle moves over the thread and forms hydrogen bonds with the ni˙− station. This process occurs on a time scale of 1 μs. Subsequently, slow (∼100 μs) charge recombination between the ni˙− station and radical cation of the electron donor occurs. This is accompanied by back-shuttling of the macrocycle. After reformation of the hydrogen bonds between the macrocycle and the succ station, the system is ready to shuttle again.2 In the time-resolved experiments we trigger the translation of the macrocycle with a short UV pulse, and observe the subsequent vibrational absorption change using a delayed mid-IR probe pulse. By recording data at different time delays of the probe pulse with respect to the pump pulse, we measure the time dependence of the absorption changes of the molecular device.
2 Experiment
2.1 Ultraviolet-pump infrared-probe setup
For the UV-IR pump-probe experiments we use a commercial Ti:sapphire laser (Spectra-Physics Hurricane, 800 μJ, 100 fs FWHM) synchronised with a commercial, pulsed Nd:YAG laser of which we use the third harmonic (IB Laser DiNY pQ, 355 nm, 0.58 mJ, 3 ns). A variable, computer-controlled time delay between the two laser outputs is achieved in a similar manner as reported by other groups,38,39 using an electronic configuration as shown in Fig. 2. A residual output from the Ti:sapphire oscillator (80 MHz) is detected by a fast photodiode, amplified and frequency-divided to produce a 1 kHz signal. This signal triggers (1) a pulse generator which provides the triggering for the YLF pump laser and Pockels-cell driver of the regenerative amplifier, (2) an electronically gated amplifier used to record the signals of the MCT-detector array and (3) a computer-controlled electronic delay generator (Berkeley Nucleonics Corporation Model 575-4C). The latter provides the triggering for the Nd:YAG laser that generates the UV pump pulse. The maximum delay between the UV and mid-IR laser pulses is determined by the repetition rate of the Ti:sapphire laser.
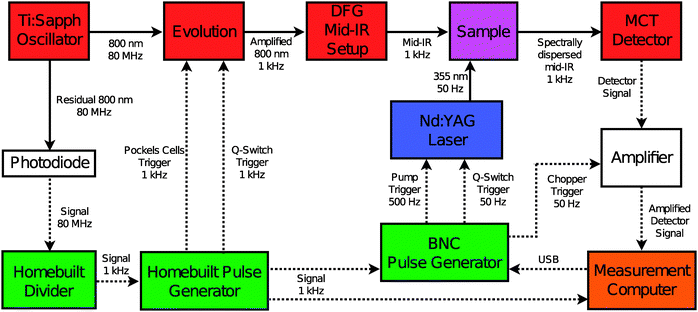 |
| | Fig. 2 Electronic setup used to synchronise the Ti:sapphire and Nd:YAG laser systems. The solid lines represent light pulses, the dashed lines represent electronic pulses. The different colors indicate: electronics (green); infrared source and detection (red); UV source (blue); sample (purple); light and signal amplification (white); data acquisition (orange). | |
The 355 nm pulse length is determined from a cross-correlation with the mid-IR pulses obtained by differentiating the pump–probe signal in a Ge plate. From a fit to a gaussian function we obtain a full-width half-maximum (FWHM) of 3.6 ± 0.4 ns for the pump pulse. The Nd:YAG laser is pumped at 500 Hz and Q-switched at 50 Hz. Using the amplified 800 nm output of the Hurricane and an optical setup described elsewhere40 we obtain mid-IR pulses with a duration of ∼150 fs, a bandwidth of 200 cm−1 and an energy of 1 μJ. Probe and reference pulses are obtained from the mid-IR light by reflection off the front and back surfaces of a wedged BaF2 window. The UV-IR pump–probe experiment is performed by focusing the 355 nm output several cm behind the sample with an f = 200 mm lens. The changes induced in the sample are monitored by the mid-IR probe pulses, which are spatially overlapped with the pump beam, at various time delays between the UV-pump and mid-IR probe pulse. The mid-IR reference beam passes through an area of the sample not influenced by the pump. Both probe and reference beams are focused through the sample by means of an f = 100 mm off-axis parabolic mirror. At the sample, the UV and mid-IR beam diameters are 1 mm and 200 μm, respectively. The polarisation of the UV-pump and mid-IR probe are perpendicular with respect to one another. Transient absorption changes are measured by frequency-dispersed detection of the mid-IR pulses using a 2 × 32 HgCdTe (MCT) array detector (Infrared Associates). Four consecutive measurements with different center frequencies of the IR probe pulse are necessary to construct the UV-IR transient spectra shown in the main text. From the overlapping spectral regions, we find that no scaling of the data of different spectral windows is required. In the case of overlapping frequencies, the pixels with the best signal-to-noise were used. All steady-state Fourier-transform infrared (FTIR) spectra are measured on a Bruker Vertex 70 spectrometer (resolution 2 cm−1). All rotaxane experiments are carried out on a solution of the rotaxane (10−4 M) and 1,4-diazabicyclo[2.2.2]octane (DABCO, 10−2 M) in CD3CN (Eurisotop, >99.8% D purity). Neither the solvent nor DABCO has absorption bands in the spectral region of interest. Argon is bubbled through the sample for 15 min in order to remove dissolved oxygen from the solution. The sample is kept in a sealed IR cell consisting of two CaF2 windows separated by a 10 mm spacer. The experiments are conducted at room temperature (21 °C).
Model calculations of the vibrational spectra in the harmonic approximation were performed on fragments of the actual rotaxanes with Gaussian09, rev. A02.41 Structures of the fragments are shown in the ESI.† For most calculations, the B3LYP hybrid functional with the 6-31G(d) basis set was used. Test calculations did not show significantly improved agreement with experiment when the 6-31+G(d) or the 6-311G(d,p) basis sets were applied. A common scaling factor of 0.973 was derived for the vibrational frequencies of all fragments by fitting them to the experimental frequencies (Table 1). The bands of the triplet state and the amide II vibrations were not used in the scaling. The scaling factor agrees well with that published by Scott and Radom.42
Table 1 Overview of the vibrational modes with their corresponding frequencies and description of the molecular shuttle in the ground, triplet and radical-anion state. “Ar” stands for aromatic ring vibration; “Rad. anion” for radical anion
| Peak number |
Mode |
Experimental frequency/cm−1 |
Calculated and scaled frequency/cm−1 |
Hydrogen-bonded |
Nature of vibration |
Electronic state of naphthalimide |
| The calculated frequencies were obtained from DFT at the B3LYP/6-31G(d) level on the following model compounds: n-Propyl-naphthalimide station; aTn-propyl-naphthalimide station in the triplet state; a˙−radical anion n-propyl-naphthalimide station. Methyl-succinamide station-macrocycle pseudo-rotaxane. Radical anion n-propyl-naphthalimide station-macrocycle pseudo-rotaxane. Methyl-succinamide station. See ESI Fig. S2 for structures of the fragments. Peaks 14 and 15 are indistinguishable in the calculations. |
| 1 |
niss
|
1700 |
1703a |
No |
Imide
|
Ground |
| 2 |
niasas
|
1663 |
1660a |
No |
Imide
|
Ground |
| 3 |
mc
|
1663 |
1664–1673b |
No |
Amide I |
Ground |
| 4 |
niArAr
|
1632 |
1627a |
No |
Ar |
Ground |
| 5 |
succ
|
1632 |
1627b |
Yes |
Amide I |
Ground |
| 6 |
niArAr
|
1603 |
1596a |
No |
Ar |
Ground |
| 7 |
niArAr
|
1580 |
1569a |
No |
Ar |
Ground |
| 8 |
succNHNH
|
1530 |
1538–1554b |
Yes |
Amide II |
Ground |
| 9 |
mcNHNH
|
1530 |
1538–1554b |
Yes |
Amide II |
Ground |
| 10 |
niss
|
1632 |
1629aT |
No |
Imide
|
Triplet |
| 11 |
niasas
|
1591 |
1584aT |
No |
Imide
|
Triplet |
| 12 |

|
1613 |
1623a˙− |
No |
Imide
|
Rad. anion |
| 13 |

|
1565 |
1571a˙− |
No |
Imide
|
Rad. anion |
| 14 |

|
1531 |
1537a˙− |
No |
Imide
|
Rad. anion |
| 15 |

|
1531 |
1537a˙− |
No |
Ar |
Rad. anion |
| 16 |
succ
|
1678 |
1689d |
No |
Amide I |
Rad. anion |
| 17 |
mc
|
1654 |
1660–1668c |
No |
Amide I |
Rad. anion |
| 18 |

|
1592 |
1581c |
Yes |
Amide I |
Rad. anion |
| 19 |
mcNHNH
|
1555 |
1554–1561c |
Yes |
Amide II |
Rad. anion |
| 20 |

|
1519 |
1516a˙− |
No |
Imide
|
Rad. anion |
| 21 |

|
1491 |
1488c |
Yes |
Mixed |
Rad. anion |
3 Results and discussion
The remainder of the article is organized as follows. We begin by discussing the steady-state IR spectra of the rotaxane and thread. After that, the transient spectra of the rotaxane as it progresses through the shuttling cycle (triplet, radical anion before shuttling, and radical anion after shuttling) are discussed. The numbering of the peaks in all spectra corresponds to that used in Table 1. The color-coding of the labels corresponds with that used for the different components of the rotaxane in Fig. 1. The complete transient spectral data are then analyzed using singular-value decomposition. By combining this analysis with a quantitative model for the kinetics, we derive the species-associated spectra of the triplet, initial radical-anion and final radical-anion states of the rotaxane and thread.
Fig. 3 shows the steady-state Fourier transform infrared (FTIR) spectra of the thread and rotaxane. The peaks in the rotaxane FTIR spectrum can be assigned by comparison with spectra of the constituent components,25 and using the results of the ab initio calculations (see ESI†). We begin by describing the infrared spectrum of the thread. The symmetric and antisymmetric CO-stretch modes of the ni station are observed at 1701 cm−1 (peak 1) and 1662 cm−1 (peak 2), respectively. The absorption band at 1662 cm−1 has a broad, high-frequency shoulder (1678 cm−1) belonging to CO-stretch vibrations of the succ station (peak 16). An aromatic ring vibration (peak 4) absorbs at 1633 cm−1. The peaks at 1605 cm−1 and 1580 cm−1 (peaks 6 and 7) are assigned to aromatic ring vibrations of the ni unit, as well. The broad band (peak 8), peaking at 1540 cm−1, is the amide II (mainly NH-bending) vibration of the succ station.
The differences between the thread and rotaxane spectra are caused by the hydrogen-bonding interaction between macrocycle and thread. These differences are most clearly observed at 1662 cm−1. Compared to the thread, the succ CO-stretch band in the rotaxane has redshifted because the CO groups of the succ station are hydrogen bonded to the macrocycle. The redshifted CO-stretch mode of the succ station appears as peak 5, which overlaps with peak 4 at 1633 cm−1. The absorption spectrum of the rotaxane also contains a contribution from the CO-stretch vibration of the macrocycle (peak 3). The final difference between the spectra of thread and rotaxane is that the amide II band in the latter contains an additional contribution from the macrocycle (peak 9). On the low-frequency side NH groups contribute that are less involved in hydrogen bonding (those of the thread), the high-frequency amide II vibrations are from hydrogen-bonded NH groups (those of the macrocycle).
3.2 Transient ultraviolet-pump infrared-probe spectra
All the transient IR spectra shown in this subsection are normalized on peak 1. The time-dependence of peak 1 is determined only by the recombination of the ni˙− station with DABCO˙+. By the normalization we remove this contribution from the signal and are left with spectral changes caused by radical-anion creation and shuttling. This makes for an easier qualitative discussion of the spectra. The recombination of the charged species is treated in Section 3.3.
3.2.1 Triplet state.
The first observable species in the UV-IR shuttling experiment is the triplet state of the ni station. This state is difficult to observe separately in the shuttling experiments, because in the presence of the external electron donor, it is rapidly converted into a radical anion (on a time scale of approximately 30 ns). We can measure the pure triplet state spectrum by exciting the rotaxane in the absence of the external electron donor. This measurement also serves as a check to confirm that the frequency shifts observed in the presence of an electron donor (Section 3) are indeed caused by reduction of the ni station. The normalized UV-IR transient spectra of the rotaxane in the absence of the electron donor are shown in Fig. 4.
 |
| | Fig. 4 Normalized UV-IR spectra of the rotaxane in the absence of an external electron donor at several delays after UV excitation. The curves are a guide to the eye. The numbering corresponds with that used in Table 1. The colors of the labels correspond to those of the components of the rotaxane shown in Fig. 1. | |
The negative peaks 1 and 2 are the ground-state bleaching of the ni symmetric and antisymmetric CO-stretch modes. This bleaching is caused by the transfer of rotaxane population from the electronic ground state to the triplet state. The absorptions of the symmetric and antisymmetric CO-stretch vibrations in the triplet state are visible at 1632 cm−1 and 1592 cm−1 (peaks 10 and 11). In the delay range probed in Fig. 4, there are no time-dependent changes in the intensities of the peaks other than the relaxation of the triplet state to the electronic ground state which occurs with a lifetime of 44 μs.43 The lack of dynamics other than the relaxation of the triplet state to the ground state suggests that no shuttling occurs in the triplet state.
3.2.2
Radical anion before shuttling.
The reduction of the ni station in the triplet state to generate a radical anion is the next step of the operating cycle. As in the case of the triplet state, the radical anion species before shuttling cannot be observed separately in the time-resolved shuttling experiment. This is because significant contributions from both triplet and final (post-shuttling) radical-anion species are always present together with the initial (pre-shuttling) radical-anion state (see Section 3.3). This problem can be solved by exciting the thread (instead of the rotaxane) in the presence of the external electron donor. The normalized UV-IR transient spectra of the thread in the presence of an electron donor are shown in Fig. 5.
 |
| | Fig. 5 Normalized UV-IR spectra of the thread in the presence of an external electron donor at different delays. The curves are a guide to the eye. The numbering corresponds with that used in Table 1. The colors of the labels correspond to those of the components of the rotaxane shown in Fig. 1. | |
At short delays, we observe the spectrum of the thread in the triplet state. This closely resembles the spectrum of the rotaxane in the absence of the external electron donor, see Fig. 4. This means that at short delays the presence or absence of the macrocycle has no influence on the transient spectrum, confirming that the spectral changes are due to changes in the naphthalimide moiety of the thread only. At delays >100 ns we observe the spectrum of the radical-anion form of the thread, generated by electron transfer from DABCO to the ni in the triplet state. A new negative signal becomes visible at 1633 cm−1 (peak 4). This is because the aryl ring vibration of the naphthalimide anion is shifted to a lower frequency with respect to the neutral species. The calculations indicate that in the anion some mixing of this aryl mode with the antisymmetric imide stretch occurs. The absorption of this mode is peak 13. The broad and intense peak found at 1531 cm−1 is another combination of the radical-anion state antisymmetric CO-stretch vibration (peak 15) and an aromatic ring vibration of the ni˙−. The ground-state bleaching of the latter is probably hidden under the low-frequency side of peak 12. The assignments of peaks 14 and 15 are based on arguments made in Section 3. In the thread no shuttling occurs, so there is no further evolution of the spectrum other than the overall decrease in signal due to charge recombination.
3.2.3
Radical anion after shuttling.
The final step in the operating cycle of the rotaxane is the formation of the final radical-anion species in which the macrocycle has broken free from the succ station, shuttled over the thread, and formed new hydrogen bonds with the ni˙− station (see Fig. 2). The spectral changes between the post-shuttling and the initial radical-anion state involve the breaking of the hydrogen bonds between the succ station and the macrocycle, and the formation of hydrogen bonds between the macrocycle and the ni˙− station. Normalised UV-IR transient spectra at several delays after UV excitation of the rotaxane in the spectral range 1480–1720 cm−1 are shown in Fig. 6.
 |
| | Fig. 6 Normalized UV-IR spectrum of the rotaxane in the spectral region of 1475–1720 cm−1. The curves are a guide to the eye. The numbering corresponds with that used in Table 1. The colors of the labels correspond to those of the components of the rotaxane shown in Fig. 1. | |
The transient spectrum of the rotaxane at 100 ns (Fig. 6) matches that of the reduced thread (Fig. 5) at delays ≥100 ns. The changes observed after 100 ns in the rotaxane spectrum can therefore be attributed to the departure of the macrocycle from the succ station and its arrival at the ni˙− station. This motion is possible due to the breaking and making of hydrogen bonds between the different components of the rotaxane involved in the shuttling. In the case of CO-stretch vibrations for this type of system, the red-shift in frequency caused by hydrogen bonding compared to the free (non-hydrogen bonded) situation is typically 20–40 cm−1.44,45 This frequency shift is accompanied by a broadening due to a distribution of possible hydrogen-bond strengths. In contrast to the CO-stretch mode, the NH-bend mode of a hydrogen bonded NH group is blue-shifted compared to that of a free NH group.44,45 The hydrogen bonding increases the restoring force of the bending mode, thereby increasing its frequency. The calculations show frequency differences of the amide II modes of up to 30 cm−1, which span the width of the observed amide II band.
The first step in the shuttling process involves the breaking of the hydrogen bonds between the macrocycle and the succ station.32 This is observed as a decrease in absorption of the hydrogen-bonded CO-stretch vibration (peak 5) as delay increases. The complementary, non-hydrogen bonded CO-stretch vibration of the succ station (peak 16) is observed at 1679 cm−1. The succ station CO-stretch frequency (peak 16) in the final state is blue-shifted compared to that in the initial state (peak 5) due to the lack of hydrogen bonding with the macrocycle. The opposite happens for the ni˙− station. In the initial state, peak 12 is free from hydrogen bonding. As time progresses, more macrocycle binds to the ni˙− station and the intensity of this peak decreases. The complementary, hydrogen bonded symmetric CO-stretch (peak 18) is observed at 1591 cm−1. Peak 18 is red-shifted compared to peak 12. This change is a direct consequence of the hydrogen bonding with the macrocycle. Peak 17 is the CO-stretch vibration of the macrocycle when it is bound to the ni˙− station. The corresponding ground-state bleaching (peak 3) is probably located under peak 2. The 9 cm−1 red shift arises because the macrocycle NH groups hydrogen-bond more strongly to the ni˙− station than to the succ station. All the CO-stretches in the rotaxane are amide I (or imide I) vibrations; they contain NH-bend character. Thus, even though the CO-groups of the macrocycle are not directly involved in the hydrogen-bond interaction, they still experience the stronger hydrogen bonding to the ni˙− station.25 The DFT calculations predict a red shift of about 5 cm−1, in good agreement with experiment.
The spectrum below 1580 cm−1 is rather congested which complicates the assignment of the peaks. However, the situation becomes more straightforward when the NH groups of the rotaxane are deuterated. All contributions from the amide II vibrations are shifted to lower frequencies, well outside our observed spectral range (see Fig. 7 top panel for the FTIR spectrum of the deuterated rotaxane). This allows us to assign the amide II modes in an unambiguous manner. A comparison between the UV-IR spectra of the normal and N-deuterated rotaxane is shown in Fig. 7.
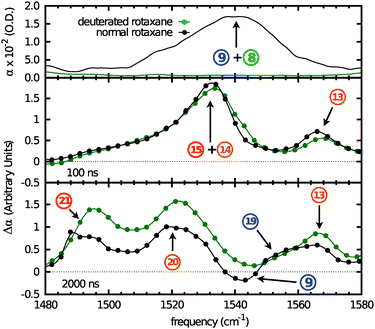 |
| | Fig. 7 Top panel: FTIR of the normal and N-deuterated rotaxane in the amide II spectral region. Center panel: normalized initial radical-anion spectra of the normal and N-deuterated rotaxanes at 100 ns after UV-excitation. Bottom panel: normalized final radical-anion spectra of the normal and N-deuterated rotaxane at 2 μs after UV-excitation. The curves are a guide to the eye. The numbering corresponds with that used in Table 1. | |
There is no difference between the transient IR spectra of the normal and deuterated rotaxane at 100 ns because there are no changes involving the amide II band between the ground- and the initial radical-anion state. Peak 9 blue-shifts upon shuttling, giving rise to a negative feature at 1540 cm−1 and a positive feature at 1555 cm−1 (peak 19). Both features disappear after N-deuteration, indicating they belong to an amide II mode. The shift to higher frequencies is evidence of increased hydrogen-bonding strength. This would be the case in the NH groups of the macrocycle. We observe that peak 13 does not change in position or intensity in the deuterated rotaxane. The shuttling of the macrocycle does not affect this peak, which indicates that it most likely belongs to an aromatic ring vibration of the ni˙− station. The broad, asymmetric absorption peaking at 1531 cm−1 at 100 ns remains after deuteration. As time progresses, it decreases in intensity and undergoes a shift towards lower frequencies. A broad peak at 1491 cm−1 (peak 21) increases in intensity with increasing delay. It is clear that the peak at 1531 cm−1 originates from several overlapping bands. The calculations (see ESI†) shed light on the nature of these absorptions: in the neutral imide, the symmetric (peak 1) and antisymmetric CO-stretching vibrations (peak 12) are clearly separated from the aromatic ring vibrations (peaks 4, 6, and 7). In the radical anion, however, the frequencies of these modes approach each other. In particular the antisymmetric CO-stretch mode (peak 15) shifts to lower frequencies and as a result mixes with an aromatic ring vibration (peak 14). In the radical anion after shuttling, a further frequency lowering leads to the mixing of the aromatic ring modes with the highest-frequency deformation mode of the tert-butyl groups. This results in a strong absorption at 1491 cm−1 (peak 21), which has no equivalent in the neutral imide or in the radical anion prior to shuttling. This also explains the shift of peak 14 to lower frequencies (peak 20). Since this predominantly aromatic ring vibration contains significant antisymmetric CO-stretch character, it will experience the hydrogen bonding of the macrocycle to the CO groups of the ni˙− station.
3.3 Shuttling dynamics
3.3.1 General considerations.
We observe that the spectral features of each of the components involved with the shuttling (succ, ni˙−, and macrocycle) show the same time-dependence.32 This can only be the case if the shuttling event in a single rotaxane occurs much faster than observed for the ensemble. The complete operation cycle of the rotaxane involves three time components (see Fig. 1): the conversion of the triplet state into the radical anion, shuttling of the macrocycle, and the charge recombination and back-shuttling. This process spans three orders of magnitude in time (see Fig. 8): the radical-anion formation is witnessed primarily within the first 100 ns. This is followed by the shuttling of the macrocycle, which occurs between 100 ns and 1 μs. Charge recombination is an ongoing process throughout the experiment. It is the main contributor to the changes in the signal from 1 μs onwards. To verify this mechanism in an objective manner, we have analysed the data using singular value decomposition.
 |
| | Fig. 8 3-Dimensional representation of the data where change in absorption (Δα) is represented as a function of frequency (cm−1) and time (ns) on a logarithmic scale. | |
3.3.2 Singular value decomposition.
There are two reasons for performing an SVD analysis of our data. The first is that such an analysis can confirm the three-stage shuttling mechanism discussed in the previous section (see also Fig. 1). This mechanism implies that at each time the transient spectrum should be a sum of the spectra of three species (triplet, radical anion before shuttling, radical anion after shuttling), with the contributions of each species depending on time. The second reason is that an SVD makes it possible to obtain the spectra of the rotaxane in each stage of the operation cycle. It is difficult to obtain these spectra directly from the transient measurements, because the shuttling does not occur synchronously for all rotaxane molecules, so that in the transient measurements one always observes a linear combination of the spectra of the individual species. In the previous sections, this problem was partly solved by “freezing” the operation cycle at a specific stage: in particular, we obtained the spectrum of the triplet state by removing the electron donor (so that no reduction occurs), and the spectrum of the pre-shuttling radical-anion state by omitting the macrocycle (so that no shuttling occurs). SVD makes it possible to extract the spectra of the different states in the operation cycle from experimental data obtained with the fully operating molecular device.
The analysis is performed as follows. Our data consist of transient spectra, measured at M frequencies and N delays, with M > N. This can be arranged in an M × N matrix D (in our case M = 77 and N = 65, see Fig. 9 for a graphical representation). This matrix can be decomposed into its principal components using SVD:33,34,46
| | 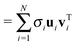 | (2) |
where
U is the
M ×
N projection
matrix, Σ the
N ×
N diagonal singular-values
matrix, and
VT the
N ×
N transposed projection
matrix. The columns of
U are the projection vectors
ui (containing
M components), also referred to as the basis spectra of the data. The rows of
VT are the target vectors
vTi (containing
N components). The elements of a specific vector
vTi represent the delay dependence of the contribution having the corresponding basis spectrum
ui, to the measured data. The data set is thus decomposed into a weighted sum of outer products
uivTi of vectors. The singular value
σi (
i = 1,2,…
N) represents the weight of the
ith component of the sum in describing the entire data set.
 |
| | Fig. 9 Singular-value decomposition of the data shown in Fig. 8. (A) 2-Dimensional representation of the data, where blue represents a negative signal and red a positive signal. Species-associated spectra (B) and time-dependence (C). The curves in panel B are guides to the eye. The curves in panel C are the fitted linear combinations of NT(t),  , and , and  . . | |
Often, there is a small subset of weights σi that are much larger than all the others. In that case, the experimental data set D is very well described by only a limited number n ≪ N of basis spectra and corresponding target vectors that have a large weight:
| |  | (3) |
where
Ũ is the reduced (truncated)
M ×
n projection
matrix,

the reduced
n ×
n singular-value
matrix, and
ṼT the reduced
n ×
N target
matrix. The discarded vectors typically contain noise contributions.
We find that our data can be well described by three vectors (n = 3 in eqn (3)). The weights and the first four spectral and target components with the largest singular values are shown in Fig. S3 and S4 (ESI†). The fourth and higher vectors all represent uncorrelated noise contributions in the signal. This was confirmed by reconstructing the data set from the truncated target, singular-value, and projection matrices. The reconstructed data set ( in eqn (3)) can be compared with the original data set (D in eqn (3)), taking the uncertainties in the experimental data points into account. The reduced chi-square of the deviations between the reconstructed and the original data obtained in this way is 3.58, which implies that the deviations are on the order of the measurement errors on the data points, and therefore not significant.
in eqn (3)) can be compared with the original data set (D in eqn (3)), taking the uncertainties in the experimental data points into account. The reduced chi-square of the deviations between the reconstructed and the original data obtained in this way is 3.58, which implies that the deviations are on the order of the measurement errors on the data points, and therefore not significant.
The target and projection vectors obtained from an SVD generally cannot be related directly to species-associated spectra and their corresponding time dependence.33,46 This is because for any invertible n × n matrix C the decomposition of eqn (3) can be rewritten as
| | 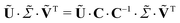 | (4) |
where the
M ×
n matrix Us ≡
Ũ·
C and the
n ×
N matrix 
contain
n alternative basis
spectra and target vectors, defined by the
n ×
n transformation
matrix C, and resulting in exactly the same reconstructed data set. Physically meaningful basis spectra (species-associated spectra) and target vectors (species-associated delay dependencies) can be obtained from the original SVD components by finding the appropriate transformation
matrix C. This is usually done by imposing constraints on the basis spectra and/or target vectors.
34 In particular, by assuming a specific kinetic model to describe the time-dependent species concentrations, it is often possible to determine
C, and therefore the species spectra, unambiguously.
To this purpose, we proceed as follows. The transformation C should be such that Us contains the spectra of the three species (triplet, radical anion before shuttling, radical anion after shuttling), and Vs the time dependencies of their concentrations. The elements of C are determined by requiring that Vs has a functional form that represents the time-dependent concentrations of the three species. This is done by assuming a quantitative kinetic model for the delay dependencies of the three species (triplet, radical anion before shuttling, radical anion after shuttling), and optimizing the agreement of Vs with the predicted time dependencies using a least-squares fit, see the next section.
3.3.3 Kinetic model.
Based on the considerations of Section 3.3, the species concentrations are determined by the following set of rate equations:| |  | (7) |
| |  | (8) |
| | 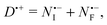 | (9) |
where NT(t) is the triplet population, kT the pseudo first-order rate constant for the triplet-to-anion conversion (see Section 3), and η the radical-anion yield (some of the population in the triplet state is lost by non-radiative decay, and part of the newly formed radical anion population is rapidly quenched after creation by DABCO˙+).37 is the initial (pre-shuttling) radical-anion population, which has three loss channels. The main loss channels are the shuttling (rate constant kS), and the charge recombination with DABCO˙+ (second-order rate constant kR). We find that a third, minor loss channel (rate constant kQ) is needed to quantitatively describe the data at long times. This contribution is probably due to quenching of the radical anion by traces of molecular oxygen present in the sample (the resulting superoxide rapidly reacts with DABCO˙+, so the oxygen concentration remains constant). This loss channel is described by the third term in eqn (7). The third rate equation represents the final (post-shuttling) radical-anion concentration
is the initial (pre-shuttling) radical-anion population, which has three loss channels. The main loss channels are the shuttling (rate constant kS), and the charge recombination with DABCO˙+ (second-order rate constant kR). We find that a third, minor loss channel (rate constant kQ) is needed to quantitatively describe the data at long times. This contribution is probably due to quenching of the radical anion by traces of molecular oxygen present in the sample (the resulting superoxide rapidly reacts with DABCO˙+, so the oxygen concentration remains constant). This loss channel is described by the third term in eqn (7). The third rate equation represents the final (post-shuttling) radical-anion concentration  , which has charge recombination and quenching as its only loss channels; finally, D˙+ is the concentration of DABCO˙+, which is determined by the requirement of conservation of charge.
, which has charge recombination and quenching as its only loss channels; finally, D˙+ is the concentration of DABCO˙+, which is determined by the requirement of conservation of charge.
Directly upon photo-excitation, intersystem crossing to the triplet occurs (the intersystem crossing can be assumed instantaneous on the time scale of the experiment). We set the initial concentration of triplet species NT = N0 (the concentration of triplet generated at t = 0) and 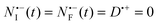 . As shown in the ESI,† for these initial values, the solutions to rate eqn (6)–(9) are given to a very good approximation by the following analytical expressions:
. As shown in the ESI,† for these initial values, the solutions to rate eqn (6)–(9) are given to a very good approximation by the following analytical expressions:
| |  | (11) |
| |  | (12) |
Given a set of rate constants kT,kS,kR, and an initial concentration N0, this set of solutions allows us to predict the species concentrations that should be contained in Vs as follows:
| |  | (13) |
where
ti are the delay values sampled in the time-resolved experiment. From previous
UV-Vis measurements it is known that
kR = 9 × 10
9 M
−1 s
−1 and
η = 0.20.
2,37 Since
C is defined such that

we can determine the unknown elements of
C and the remaining unknown parameters
N0,
kT, and
kR from a global least-squares fit of
C·
Vcalcs to the matrix
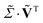
as obtained from the SVD. From the resulting least-squares fit, we find
kS = 0.0013 ns
−1,
kT = 0.04 ns
−1, and
N0 = 5.8 × 10
−5. These values agree well with the ones obtained previously using a smaller data set.
32 Additionally, we find
kQ = 8.8 × 10
−5ns
−1.
The species-associated spectra and corresponding time-dependences can now be generated from the resulting C matrix. They are shown in Fig. 9 (panels B and C, respectively). The species-associated spectra are in good agreement with the spectra obtained by “freezing” the operation cycle: for the triplet spectrum, see Fig. 4; for the pre-shuttling radical anion, see Fig. 5 at delays ≥100 ns; for the post-shuttling radical anion, see Fig. 6 at delays ≥2 μs. Fig. 9C shows clearly that during the shuttling cycle, most of the time more than one species is present in the sample. In particular, the only species that can be observed in an isolated manner is the radical anion after shuttling, which is the only species present for t > 5 μs. As stated before, the advantage of the SVD method is that the species-associated spectra can be determined from the convoluted data set in an unambiguous manner, provided that the kinetic scheme of the dynamic process being studied is known.
4 Conclusion
The work presented here is an extension to the work we published previously on the operation mechanism of a [2]rotaxane.32 We have extended the range of measured frequencies of the transient IR spectra to lower energies, and comparison of the transient spectra of the normal and N-deuterated rotaxane has enabled us to assign the convoluted peaks observed in the extended spectral range. The positions and shifts of the peaks are confirmed by DFT calculations performed at the B3LYP/6-31G(d) level. Understanding the assignment and the shifts of the low-frequency range opens up new possibilities for future work. We can study, for example, the shuttling motion of the rotaxane in solvents that are not transparent in the amide I frequency range but are so in the amide II range. Also, since the modes in the amide II frequency range are of a different nature than those in the amide I range, effects involving different functional groups of the rotaxane (in particular NH or CO groups) can be observed separately.
We have used singular value decomposition to obtain the number of species involved in the operation cycle in an objective manner. We applied a kinetic model to the SVD that takes the time-dependence of all three photochemical species into account. In this way we obtained the rates of the different stages of the rotaxane operating cycle as well as the species associated spectra. The SVD allows us to use the entire data set (both the full frequency and the delay ranges) which results in a much more precise determination of the rates involved in the shuttling process than was achieved in our earlier studies. Finally, from the SVD we can determine the spectra of the intermediate species without having to “freeze” the operation cycle of the molecular device.
Acknowledgements
We thank Chris N. van Dijk for valuable discussions. We thank Paul Reinders, Steven Kettelarij, Dick Bebelaar, Michiel Groeneveld, Henk Luiten, Harry Beukers, and Cees van den Biggelaar for technical support. This work was supported by the European Community (EMMMA IHP Research Training Network, contract HPRN-CT-2002-00168, and Hy3M STREP, contract NMP4-CT-2004-013525) and by the Stichting voor Fundamenteel Onderzoek der Materie (FOM) which is financially supported by the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO).
References
- Wolfram Research, Inc., Mathematica Edition: Version 6.0, 2007.
- A. M. Brouwer, C. Frochot, F. G. Gatti, D. A. Leigh, L. Mottier, F. Paolucci, S. Roffia and G. W. H. Wurpel, Science, 2001, 291, 2124–2128 CrossRef CAS.
- A. Altieri, F. Gatti, E. R. Kay, D. A. Leigh, D. Martel, F. P. A. M. Slawin and J. K. Wong, J. Am. Chem. Soc., 2003, 125, 8644–8654 CrossRef CAS.
- D. D. Gunbas, L. Zalewski and A. M. Brouwer, Chem. Commun., 2010, 46, 2061–2063 RSC.
- K. Mislow, Acc. Chem. Res., 1976, 9, 26–33 CrossRef CAS.
- H. Iwamura and K. Mislow, Acc. Chem. Res., 1988, 21, 175–182 CrossRef CAS.
- V. Serreli, C. F. Lee, E. R. Kay and D. A. Leigh, Nature, 2007, 445, 523–527 CrossRef CAS.
- T. D. Nguyen, Y. Liu, S. Saha, K. C. F. Leung, J. F. Stoddart and J. L. Zink, J. Am. Chem. Soc., 2007, 129, 626–634 CrossRef CAS.
- T. C. Bedard and J. S. Moore, J. Am. Chem. Soc., 1995, 117, 10662–10671 CrossRef CAS.
- T. R. Kelly, H. De Silva and R. A. Silva, J. Am. Chem. Soc., 1999, 401, 150–152 CAS.
- N. Koumura, R. W. J. Zijlstra, R. A. van Delden, N. Harada and B. L. Feringa, Nature, 1999, 401, 152–155 CrossRef CAS.
- D. A. Leigh, J. K. Y. Wong, F. Dehez and F. Zerbetto, Nature, 2003, 424, 174–179 CrossRef CAS.
- J. V. Hernández, E. R. Kay and D. A. Leigh, Science, 2004, 306, 1532–1537 CrossRef CAS.
- T. R. Kelly, M. C. Bowyer, V. Bhaskar, D. Bebbington, A. Garcia, F. Lang, M. H. Kim and M. P. Jette, J. Am. Chem. Soc., 1994, 116, 3657–3658 CrossRef CAS.
- J. D. Badjić, V. Balzani, A. Credi, S. Silvi and J. F. Stoddart, Science, 2004, 303, 1845–1849 CrossRef CAS.
-
R. Kubo and N. Hashitsume, Statistical Physics II: Nonequilibrium Statistical Mechanics, Springer, Berlin, 1985 Search PubMed.
- R. D. Astumian, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 1843–1847 CrossRef CAS.
- R. D. Astumian, Phys. Chem. Chem. Phys., 2007, 9, 5067–5083 RSC.
- R. D. Astumian, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 19715–19718 CrossRef CAS.
- K. Nørgaard, B. W. Laursen, S. Nygaard, K. Kjaer, H.-R. Tseng, A. H. Flood, J. F. Stoddart and T. Bjørnholm, Angew. Chem., Int. Ed., 2005, 44, 7035–7039 CrossRef.
- S. S. Zhu, P. J. Carrol and T. M. Swager, J. Am. Chem. Soc., 1996, 118, 8713–8714 CrossRef CAS.
- S. Garaudée, S. Silvi, M. Venturi, A. Credi, A. H. Flood and J. F. Stoddart, ChemPhysChem, 2005, 6, 2145–2152 CrossRef CAS.
- D. S. Marlin, D. González Cabrera, D. A. Leigh and A. M. Z. Slawin, Angew. Chem., Int. Ed., 2006, 45, 77–83 CrossRef CAS.
- S. Saha, A. H. Flood, J. F. Stoddart, S. Impellizzeri, S. Slivi, M. Venturi and A. Credi, J. Am. Chem. Soc., 2007, 129, 12159–12171 CrossRef CAS.
- D. C. Jagesar, F. Hartl, W. J. Buma and A. M. Brouwer, Chem.–Eur. J., 2008, 14, 1935–1946 CrossRef CAS.
- F. G. Gatti, S. León, J. K. Y. Wong, G. Bottari, A. Altieri, M. A. F. Morales, S. J. Teat, C. Frochot, D. A. Leigh, A. M. Brouwer and F. Zerbetto, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10–14 CrossRef CAS.
- K. Kwac and M. Cho, J. Chem. Phys., 2003, 119, 2247–2255 CrossRef CAS.
- C. Kolano, J. Helbing, M. Kozinski, W. Sander and P. Hamm, Nature, 2006, 444, 469–472 CrossRef CAS.
- P. Bodis, M. R. Panman, B. H. Bakker, A. Mateo-Alonso, M. Prato, W. J. Buma, A. M. Brouwer, E. R. Kay, D. A. Leigh and S. Woutersen, Acc. Chem. Res., 2009, 49, 1462–1469 CrossRef.
- A. Pfeifer, T. Majerus, K. Zikihara, D. Matsuoka, S. Tokutomi, J. Heberle and T. Kottke, Biophys. J., 2009, 96, 1462–1470 CrossRef CAS.
- J. Bredenbeck, J. Helbing, R. Behrendt, C. Renner, L. Moroder, J. Wachtveitl and P. Hamm, J. Phys. Chem. B, 2003, 107, 8654–8660 CrossRef CAS.
- M. R. Panman, P. Bodis, D. J. Shaw, B. H. Bakker, A. C. Newton, E. R. Kay, D. A. Leigh, A. M. Brouwer, W. J. Buma and S. Woutersen, Science, 2010, 328, 1255–1258 CrossRef CAS.
- E. R. Henry, Biophys. J., 1997, 72, 652–673 CrossRef CAS.
- M. Kubista, J. Nygren, A. Elbergali and R. Sjöback, Crit. Rev. Anal. Chem., 1999, 29, 1–28 CAS.
- H. L. Frisch and E. Wasserman, J. Am. Chem. Soc., 1961, 83, 3789–3795 CrossRef CAS.
- V. Wintgens, P. Valat and J. Kossanyl, J. Chem. Soc. Faraday Trans., 1994, 90, 411–421 RSC.
- D. C. Jagesar, S. M. Fazio, J. Taybi, E. Eiser, F. G. Gatti, D. A. Leigh and A. M. Brouwer, Adv. Funct. Mater., 2009, 19, 3440–3449 CrossRef CAS.
- H. S. Chung, M. Khalil, A. W. Smith and A. Tokmakoff, Rev. Sci. Instrum., 2007, 78, 063101 CrossRef.
- C. Krejtschi and K. Hauser, Eur. Biophys. J., 2011, 40, 673–685 CrossRef CAS.
- P. Bodis, O. F. A. Larsen and S. Woutersen, J. Phys. Chem. A, 2005, 109, 5303–5306 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09 (Revision A.02), Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
- A. P. Scott and L. Radom, J. Phys. Chem., 1996, 100, 16502–16513 CrossRef CAS.
-
G. W. H. Wurpel, PhD thesis, University of Amsterdam, 2001 Search PubMed.
- W. Reckien, B. Kirchner and S. D. Peyerimhoff, J. Phys. Chem. A, 2006, 110, 12963–12970 CrossRef CAS.
- A. M. Rijs, I. Compagnon, J. Oomens, J. S. Hannam, D. A. Leigh and W. J. Buma, J. Am. Chem. Soc., 2009, 131, 2428–2429 CrossRef CAS.
-
W. H. Press, S. A. Teukolsky, W. T. Vetterling and B. P. Flannery, Numerical Recipes in C, Cambridge University Press, Cambridge, UK, 1992 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Cross-correlation of the UV pump pulse with the mid-IR probe pulse (Fig. S1); structures of the model systems used for the calculations of the vibrations (Fig. S2); plot of the weights of the principal components obtained from the SVD (Fig. S3); plots of the first four target and projection vectors obtained from the SVD (Fig. S4); the Mathematica1 code for the numerical integration of the kinetic differential equations and their proposed solutions. See DOI: 10.1039/c1cp22146a |
|
| This journal is © the Owner Societies 2012 |
Click here to see how this site uses Cookies. View our privacy policy here. ![Chemical structures of the [2]rotaxane shuttle in the neutral, initial radical-anion and final radical-anion states. 1,4-Diazabicyclo[2.2.2]octane was used as external electron donor.](/image/article/2012/CP/c1cp22146a/c1cp22146a-f1.gif)
