A novel benzene–water azeotrope route to new Na-based metal fluorosulphates NaFeSO4F and NaFeSO4F·2H2O in one minute†
Zhengqiu
Yuan
,
Denghu
Wei
,
Yan
Wang
,
Yongchun
Zhu
*,
Yitai
Qian
* and
Kaibing
Tang
Department of Chemistry, University of Science and Technology of China, Hefei, 230026, P. R. China. E-mail: yuanzhengqiu@163.com; Fax: + 86-551-3607402; Tel: + 86-551-3601589
First published on 2nd March 2012
Abstract
A one-step benzene–water azeotrope method has been carried out for the first time for the selective preparation of NaFeSO4F and NaFeSO4F·2H2O starting from FeSO4·7H2O and NaF in a short time (1 min) at a low temperature (200 °C) in an autoclave with different sealed caps using benzene as a reaction medium and dehydrant.
Rechargeable Li-ion batteries have been highlighted as major power options for the “green” energy storage systems.1–4 Olivine-type LiFePO4 as a Li-ion battery cathode material has attracted much attention due to its extremely high structural stability, dependable safety, potentially low production cost and high theoretical capacity (∼170 mAh g−1, 3.4 V vs. Li).5–8 However, LiFePO4 is unable to provide sufficient voltage. In the search for new lithium-containing oxianion cathode materials with Fe(II), recent studies have focused on changing anions; fluorinated iron sulfate LiFe(II)SO4F9 (140 mAh g−1, 3.6 V vs. Li) is designed and prepared by ionothermal techniques with expensive ionic liquids. The isostructural replacement of (PO4)3− with (SO4)2− in Fe-based NASICON-type structures can improve the redox voltage by 800 mV.10,11 Additionally, the introduction of fluorine can improve the open circuit voltage and redox potential of these systems.12 The Na-based compound NaFeSO4F which also presents some redox activity (Fe3+/Fe2+) toward Li at 3.6 V has been prepared at 300 °C for 9 h using a two-step route in an ionic liquid medium.13 Other two-step methods have been used to prepare NaFeSO4F via solid-state13 (300 °C, 40–50 h) and wet chemistry routes such as the hydrophilic tetraethylene glycol method14 (220 °C, 48 h). However, these reported methods require a long reaction time (∼50 h) to obtain a high crystallinity product and are all two-step processes. This includes the preparation of the precursor FeSO4·H2O which is considered to be essential in the preparation of NaFeSO4F in the first step, as these reactions have been shown to be topotactic with the replacement of O2− from H2O by F− in FeSO4·H2O together with the ingress of one Na+ for charge compensation.
Recently, a new family of fluorosulphates NaMSO4F·2H2O (M = Fe, Co, and Ni) are prepared from MSO4·nH2O (∼5 h) and NaF in aqueous solution.12 However, among these new compounds, it is reported that NaFeSO4F·2H2O which is prepared under nitrogen is the most difficult compound to obtain because of the tendency of Fe2+ (aq) to oxidize to Fe3+ (aq) in an oxygen containing environment like common deionized water.
Can NaFeSO4F be obtained from FeSO4·7H2O directly without the preparation of FeSO4·H2O? Can the reaction of preparing NaFeSO4F be finished in a short time? It is considered that the reaction to prepare NaFeSO4F from FeSO4·7H2O and NaF involves two simultaneous processes: 1) dehydration of FeSO4·7H2O and 2) insertion of NaF in the structure and the substitution of F for O. FeSO4 will be obtained if the rate of step 1 is much higher than the step 2.14 On the contrary, NaFeSO4F·nH2O may be prepared if the rate of step 2 is higher than step 1, such as NaFeSO4F·2H2O. In the structure of FeSO4·7H2O (see Fig. S1, ESI†), the FeO6 octahedron and SO4 tetrahedron are separate; there is no shared atom between them. Two of the six oxygen atoms in the FeO6 octahedron are the bridges of two octahedrons in FeSO4·H2O (Fig. S2, ESI†), another four are distributed in four different SO4 tetrahedrons as the shared atoms. So the structure of FeSO4·7H2O is looser than FeSO4·H2O. We also can get the result from the lattice parameters (FeSO4·7H2O, a = 14.07 Å, b = 6.51 Å, c = 11.04 Å, JCPDS No. 76-0657; FeSO4·H2O, a = 7.075 Å, b = 7.541 Å, c = 7.600 Å, JCPDS No. 74-1332): the distance between the FeO6 octahedron and SO4 tetrahedron in FeSO4·7H2O is bigger than in FeSO4·H2O. The rate of insertion of NaF can be increased obviously when the raw material is FeSO4·7H2O because the resistance of NaF inserting into FeSO4·7H2O is much smaller than inserting into the tight FeSO4·H2O based on the analysis of the structures. If NaFeSO4F could be prepared from FeSO4·7H2O, the reaction time will be much shorter than preparation from FeSO4·H2O. Benzene which can form a benzene–water azeotrope with a little bit of water and can not dissolve NaFeSO4F is selected as the reaction medium and dehydrant because it can absorb the crystallization water from FeSO4·7H2O into itself to form the azeotrope to accelerate the rate of dehydration and promote the chemical equilibrium to the product.
In this communication, we report a one-step benzene–water azeotrope route to NaFeSO4F and NaFeSO4F·2H2O starting from FeSO4·7H2O and NaF directly in 1 min at 200 °C. NaFeSO4F can be obtained in an autoclave with a sealed iron cap. Interestingly NaFeSO4F·2H2O is obtained when substituting a copper cap for an iron cap at the same reaction conditions. Furthermore, using the autoclave with a copper cap, the transformation from NaFeSO4F·2H2O to NaFeSO4F can be achieved when prolonging the reaction time from 1 min to 40 h. It is noted that the tendency of Fe2+ to oxidize to Fe3+ can be effectively avoided in our sealed synthetic system of preparing NaFeSO4F and NaFeSO4F·2H2O. In addition, we have extended this one-step benzene–water azeotrope route to the preparation of NaMSO4F·2H2O (M = Co, Ni). Please refer to the ESI for experimental details.†Fig. 1 shows the X-ray powder diffraction (XRD) patterns of the products prepared in the autoclave using an iron cap (a) and a copper cap (b), respectively. The position and the relative intensity of all diffraction peaks in Fig. 1a are consistent with monoclinic NaFeSO4F (SG = C2/c). All diffraction peaks can be indexed and the lattice parameters can be calculated as a = 6.6797(4) Å, b = 8.7127(8) Å, c = 7.1910(2) Å, β = 113.493(0)°, V = 383.82(0) Å3 by Program Crysfire. All the peaks in Fig. 1b can be indexed to monoclinic NaFeSO4F·2H2O (SG = P21/m) with the calculated lattice parameters of a = 5.7338(2) Å, b = 7.3929(9) Å, c = 7.2145(6) Å, β = 112.958(8)°, V = 281.60(2) Å3.
 | ||
| Fig. 1 X-Ray powder diffraction patterns with the experimental data shown for (a) NaFeSO4F (λCu): C2/c, a = 6.6797(4) Å, b = 8.7127(8) Å, c = 7.1910(2) Å, β = 113.493(0)°, V = 383.82(0) Å3; (b) NaFeSO4F·2H2O (λCu): P21/m, a = 5.7338(2) Å, b = 7.3929(9) Å, c = 7.2145(6) Å, β = 112.958(8)°, V = 281.60(2) Å3. | ||
For NaFeSO4F, the TEM study reveals particle sizes ranging from 0.3–0.5 μm (Fig. 2a), and the corresponding SAED pattern shows an array of parallelogram symmetry dots, which is indexed using [3–12]* zone axis (Fig. 2b). For NaFeSO4F·2H2O, the TEM study reveals particle sizes ranging from 0.3–0.5 μm (Fig. 2c), and the corresponding SAED pattern also shows an array of parallelogram symmetry dots, which is indexed using [1–10]* zone axis (Fig. 2d). From the XPS spectra collected on the NaFeSO4F and NaFeSO4F·2H2O, the molar ratio of Na![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe:S:O:F is close to 1:1:1:4:1 in accord with the chemical formula of the NaFeSO4F (Fig. S3a, ESI†) and the molar ratio of Na:Fe:S:O:F is close to 1:1:1:6:1 in NaFeSO4F·2H2O (Fig. S3b, ESI†).
:Fe:S:O:F is close to 1:1:1:4:1 in accord with the chemical formula of the NaFeSO4F (Fig. S3a, ESI†) and the molar ratio of Na:Fe:S:O:F is close to 1:1:1:6:1 in NaFeSO4F·2H2O (Fig. S3b, ESI†).
 | ||
| Fig. 2 TEM images of benzene–water azeotrope synthesized (a) NaFeSO4F and (c) NaFeSO4F·2H2O. Corresponding SAED patterns of (b) NaFeSO4F and (d) NaFeSO4F·2H2O. | ||
When substituting a copper cap for an iron cap in the synthesis process, the formation of NaFeSO4F·2H2O may be due to the copper slowing down the rate of dehydration. To reveal the role of the copper, a control experiment is carried out by treating the raw materials FeSO4·7H2O and NaF in the autoclave with an iron cap at 200 °C for 1 min when a piece of copper is present in the reaction system, and NaFeSO4F·2H2O can also be obtained. Using the autoclave with a sealed copper cap, the transformation from NaFeSO4F·2H2O to NaFeSO4F can be realized when prolonging the reaction time from 1 min to 40 h. Fig. S4 shows the XRD patterns of the products obtained with different reaction times. After 2h, the main products are still NaFeSO4F·2H2O, while a small amount of NaFeSO4F appeared. After 20 h, the products are mainly NaFeSO4F coexisting with a small amount of NaFeSO4F·2H2O. Only single phase NaFeSO4F can be detected after 40 h. It is speculated that the crystallization water of NaMSO4F·2H2O can be absorbed by benzene to form NaMSO4F. There is no other obvious impurity in the transformation of NaFeSO4F·2H2O to NaFeSO4F. The mechanism of this transformation is similar to that of NaMSO4F·2H2O12 in ionic liquid.
The NaFeSO4F synthetic mechanism of our method is different from the ones beginning with FeSO4·H2O and NaF reported before. We want to explain the possible synthesis mechanism in our method. We assume two possible reaction intermediates FeSO4·H2O and NaFeSO4F·2H2O as the actual intermediate is difficult to be detected. The FeSO4·H2O, which is prepared by using the benzene in the autoclave with the iron cap at 200 °C for 1 min or by a ceramic route14, and NaF are stable and there is no new product at 200 °C in our synthetic system. Additionally, the in situ preparation of these intermediates and their subsequent conversion to the final product NaFeSO4F may be a completely different reaction to the preparation of these intermediates ex-situ and then reacting them, because of the presence of extra molecules of water in the system. For example, in situ, FeSO4·H2O will be formed only after the loss of six molecules of water from the precursor FeSO4·7H2O. The presence of this extra water may partially dissolve NaF and make it more reactive to FeSO4·H2O, thus eventually forming NaFeSO4F. The experiment reacting the FeSO4·H2O and NaF in the same reaction conditions with little water in 1 min based on the above consideration gives no new product. When we take NaFeSO4F·2H2O as the raw material into the autoclave with an iron cap, the NaFeSO4F is obtained at 200 °C for 4 h which is much longer than 1 min. These experiments reveal that the reaction intermediate may be neither FeSO4·H2O nor NaFeSO4F·2H2O. Here, we assume an idealized reaction model to tentatively explain the possible synthesis mechanism using the structural relationship between the crystal structure of the reactant and the product. There are six coordinated water molecules and one free water molecule in the crystal structure of FeSO4·7H2O (Fig. S1, ESI†). It is known that the free water molecule escapes easily from the structure (see Fig. 3a) at the beginning of the reaction; at the same time Na+–F− rapidly occupy the vacancy of the leaving water to form the ideal intermediate NaFeSO4F·6H2O (see Fig. 3a, NaFeSO4F·6H2O which has not yet been reported but is proposed for explaining the reaction mechanism). The free water molecule H+–OH− is replaced by Na+–F−. Then the Na+–F− participates in the dehydration process. With the increasing reaction time and temperature, the two coordinated waters (area I, Fig. 3b) are escaping from the intermediate in the formation of free water, in the meantime, the nearest F− gradually occupies the site of the two coordinated waters to connect the two FeF(H2O)5 octahedrons as the shared atom and the FeF2(H2O)4 octahedron chains are formed in this way (see Fig. 3c). The rest of the four coordinated waters (area II, Fig. 3b) in the FeF2(H2O)4 interact with the oxygen atoms of the different four SO4 tetrahedrons nearby to eliminate four hydrones (see Fig. 3c); the FeO4F2 octahedron is connected to four SO4 tetrahedrons with the four shared oxygen atoms. At last, the NaFeSO4F (see Fig. 3d) is obtained. All presumed reaction processes are shown in Fig. 3. The study of the actual reaction mechanism of our synthesis system is now in process.
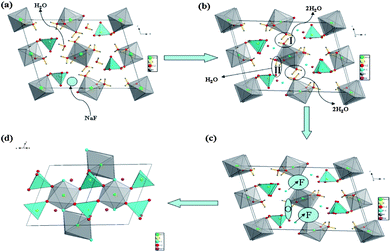 | ||
| Fig. 3 The idealized model of preparing the NaFeSO4F from the raw material FeSO4·7H2O and NaF: (a) initial FeSO4·7H2O; (b), (c) the ideal intermediate NaFeSO4F·6H2O structure; (d) the final NaFeSO4F structure. | ||
The one-step benzene–water azeotrope route to NaFeSO4F·2H2O using the autoclave with a copper cap can also be extended to synthesize NaMSO4F·2H2O (M = Co, Ni). Fig. S5 shows the X-ray powder diffraction pattern with the experimental data shown for (a) NaCoSO4F·2H2O (λCu): P21/m, a = 5.7317(1) Å, b = 7.3162(3) Å, c = 7.1918(3) Å, β = 113.53(0)°, V = 276.50(9) Å3; (b) NaNiSO4F·2H2O (λCu): P21/m, a = 5.6873(2) Å, b = 7.2583(7) Å, c = 7.1305(2) Å, β = 112.697(3)°, V = 269.34(8) Å3. We also have studied the thermostability and XPS of NaMSO4F·2H2O (M = Co, Ni) prepared by our method. The details are shown in the ESI (Fig. S6–S7, ESI†).
The NaFeSO4F and NaFeSO4F·2H2O are tested in an electrochemical cell vs. a metal Li anode to check their electrochemical activities. The results of electrochemical cycling of NaFeSO4F in a Li-electrolyte cell cycled between 2.5 V and 5.0 V vs. Li/Li+ at a rate of C/20 (the current density of charge–discharge, 1 C = 137 mA g−1) are shown in Fig. 4a. Upon discharge, a plateau near 3.6 V toward Li is observed. Though NaFeSO4F has a theoretical of capacity of 137 mAh g−1, we could hardly achieve ∼7% capacity, which is similar to the result13 (∼6% capacity) reported before. For the NaFeSO4F·2H2O, no electrochemical activity is observed (Fig. 4b); the result is consistent with the previous report.12
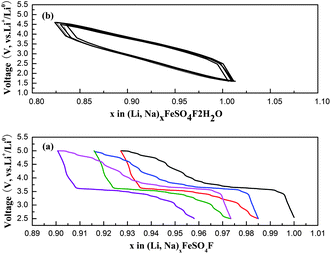 | ||
| Fig. 4 (a) Electrochemical voltage profile of NaFeSO4F cycled in a lithium cell with metallic Li anode at a rate of C/20. (b) Electrochemical voltage profile of NaFeSO4F·2H2O cycled in a lithium with metallic Li anode at a rate of C/20. | ||
In summary, we have demonstrated a novel one-step benzene–water azeotrope approach for the selective preparation of NaFeSO4F and NaFeSO4F·2H2O starting from FeSO4·7H2O in a short reaction time 1 min at the low temperature 200 °C using the autoclave with a sealed iron-cap and copper-cap. The transformation which was achieved in the ionic liquid of NaFeSO4F·2H2O to NaFeSO4F has also been achieved in benzene. The use of FeSO4·7H2O and the solvent benzene is favorable for the rapid synthesis of NaFeSO4F and NaFeSO4F·2H2O. Considering 1 mol NaF rapidly diffusing into 1 mol FeSO4·7H2O which has a looser structure, can 2 mol or 3 mol NaF diffuse into FeSO4·7H2O and react with the parent structure to form new Na-based fluorosulphates? The method of preparing NaFeSO4F·2H2O has been successfully extended to NaMSO4F·2H2O (M = Co, Ni). This communication may introduce a new idea for the rapid synthesis of other new fluorosulphates in one step, such as LiZnSO4F15 or NaZnSO4F which is unstable in aqueous solution, from the sulfate containing crystallization water.
This work is financially supported by the 973 Project of China (No. 2011CB935900).
Notes and references
- S. Nishimura, G. Kobayashi, K. Ohoyama, R. Kanno, M. Yashima and A. Yamada, Nat. Mater., 2008, 7, 707–711 CrossRef CAS.
- K. Kang, Y. S. Meng, J. Breger, C. P. Grey and G. Ceder, Science, 2006, 311, 977–980 CrossRef CAS.
- H. S. Fang, L. P. Li, Y. Yang, G. F. Yan and G. S. Li, Chem. Commun., 2008, 1118–1120 RSC.
- M. S. Whittingham, Science, 1976, 192, 1126–1127 CAS.
- S. Y. Chung, J. T. Bloking and Y. M. Chiang, Nat. Mater., 2002, 1, 123–128 CrossRef CAS.
- L. Laffont, C. Delacourt, P. Gibot, M. Y. Wu, P. Kooyman, C. Masquelier and J. M. Tarascon, Chem. Mater., 2006, 18, 5520–5529 CrossRef CAS.
- H. Gwon, D.-H. Seo, S.-W. Kim, J. Kim and K. Kang, Adv. Funct. Mater., 2009, 19, 3285 CrossRef CAS.
- Jongsoon Kim, Dong-Hwa Seo, Sung-Wook Kim, Young-Uk Park and Kisuk Kang, Chem. Commun., 2010, 46, 1305–1307 RSC.
- N. Recham, J.-N. Chotard, L. Dupont, C. Delacourt, W. Walker, M. Armand and J.-M. Tarascon, Nat. Mater., 2010, 9, 68–74 CrossRef CAS.
- A. K. Pahdi, M. Manivannan and J. B. Goodenough, J. Electrochem. Soc., 1998, 145, 1518–1520 CrossRef.
- A. K. Pahdi, K. S. Nanjundasvamy, C. Masquelier and J. B. Goodenough, J. Electrochem. Soc., 1997, 144, 2581–2586 CrossRef.
- M. Ati, L. Dupont, N. Recham, J. N. Chotard, W. T. Walker, C. Davoisne, P. Barpanda, V. Sarou-Kanian, M. Armand and J. M. Tarascon, Chem. Mater., 2010, 22, 4062–4068 CrossRef CAS.
- Prabeer Barpanda, Jean-No€el Chotard, Nadir Recham, Charles Delacourt, Mohamed Ati, Loic Dupont, Michel Armand and Jean-Marie Tarascon, Inorg. Chem., 2010, 49, 7401–7413 CrossRef CAS.
- Rajesh Tripathi, T. N. Ramesh, Brian L. Ellis and Linda F. Nazar, Angew. Chem., Int. Ed., 2010, 49, 8738–8742 CrossRef CAS.
- Prabeer Barpanda, Jean- Noel Chotard, Michael Deschamps and Jean-Marie Tarascon, Angew. Chem., 2011, 123, 2574–2579 CrossRef.
Footnote |
| † Electronic Supplementary Information (ESI) available: synthesis, crystal structure, XPS spectra, electrode preparation and testing. See DOI: 10.1039/c2ce25155h/ |
| This journal is © The Royal Society of Chemistry 2012 |