2H-perovskite related oxides: Synthesis, structures, and predictions
Hans-Conrad
zur Loye
*,
Qingbiao
Zhao
,
Daniel E.
Bugaris
and
W. Michael
Chance
Department of Chemistry & Biochemistry, University of South Carolina, Columbia, SC 29208, USA
First published on 4th November 2011
Abstract
The ability to synthesize new complex oxide materials that belong to any of the large number of known oxide structural families relies typically on a general understanding of the relationship between the specific structure type and the chemical composition of its members. However, before one can create such a structure-composition relationship that enables the synthesis of new members, one needs structural information about a sizable number of existing compositions belonging to this structural family, somewhat of a “chicken or the egg” problem. In this Highlight we will use one family of oxides, specifically oxides related to the hexagonal perovskite structure, to illustrate how exploratory crystal growth methods have been used successfully to synthesize enough diverse compositions to enable the formulation of a general structural description. Furthermore, by now it appears that enough members with different compositions have been synthesized so that one can attempt to create a structure-composition relationship that has predictive powers.
 Hans-Conrad zur Loye | Dr Hans-Conrad zur Loye is the David W. Robinson Palmetto Professor in the Department of Chemistry and Biochemistry at the University of South Carolina. He received his Bachelor of Science Degree at Brown University in 1983 and his Ph.D. in Chemistry from the University of California, Berkeley in 1988 under the supervision of Dr A. Stacy. He spent one year as a postdoctoral fellow at Northwestern University in the group of Dr D. Shriver, before starting as an assistant professor in the Chemistry Department of MIT in 1989. In 1996 he moved to the University of South Carolina where his research interests focus on the area of inorganic materials chemistry, in particular the synthesis of inorganic/organic hybrid materials and the crystal growth of new oxide materials. |
 Qingbiao Zhao | Qingbiao Zhao graduated from University of Science & Technology of China with a B.S. in Chemistry in 2006. In August 2007, he came to University of South Carolina (USA) to pursue a Ph.D. degree in Chemistry under the advisement of Dr Hans-Conrad zur Loye. He is expecting to graduate in August 2011. His research interests focus on inorganic material chemistry, including the synthesis of novel oxide materials and the relationship between crystal structures and physical properties. |
 Daniel E. Bugaris | Daniel E. Bugaris graduated with a B.S. in Chemistry from the University of Notre Dame (USA) in 2005, and a Ph.D. in Chemistry from Northwestern University (USA) in 2009. He is currently a post-doctoral fellow in the laboratory of Dr Hans-Conrad zur Loye at the University of South Carolina (USA). His research interest focuses on inorganic materials chemistry, including the crystal growth of novel compositions (oxides and chalcogenides), structure determinations, and property measurements. |
 W. Michael Chance | W. Michael Chance completed his undergraduate studies at Murray State University in 2007. He then worked on the quantum mechanical calculation-based prediction of lipid peroxidation of chloropropanes under the supervision of Dr Ricky Cox. Michael began his Ph.D. studies under Dr Hans-Conrad zur Loye at the University of South Carolina in August of 2009 where his current focus is synthesis of novel mixed-metal oxides via hydrothermal synthesis, spray pyrolysis, and a hydroxide precursor method. |
1. Introduction
The ABO3 perovskite family of oxides has been studied extensively because of the diverse properties exhibited by different elemental compositions that form in this structure type, including complex magnetic phenomena, superconductivity, ferroelectricity, and ionic conductivity to mention a few, while other researchers have explored the synthesis of new compositions. As a result, different approaches to describe the structure have been developed over the years to support the specific aspect one wishes to emphasize.1 For example, the ideal cubic perovskite structure can be described as consisting of corner-sharing BO6 octahedra with the A cation occupying the 12-fold coordination site in the middle of a cube formed by 8 such BO6 octahedra. Numerous structural variants exist due to size limitations between the A and the B cation that lead to the formation of distinct tilting modes of the octahedra if A is “too small” for B, or to the formation of the hexagonal variant consisting of infinite chains of face-sharing BO6 octahedra if A is “too large” for B.An alternative approach to describing both the cubic and the hexagonal perovskite structures is based on the stacking of close-packed [AO3] layers and the subsequent filling of the generated octahedral sites by the B cation.2 An ABC stacking sequence of [AO3] layers results in the cubic perovskite structure, while an AB stacking sequence of [AO3] layers results in the hexagonal (2H) perovskite structure. An intriguing variant of the 2H-perovskite structure was discovered by Randall and Katz when they prepared Sr4PtO6,3 which contains trigonal prisms in addition to octahedra in the chains. This was the first example of a large series of oxides referred to as 2H-perovskite related oxides. In this Highlight, we will focus on the crystal growth and crystal structures of this family of oxides, limiting ourselves to those compositions that have appeared since the first review of this structural family by Stitzer et al.,4 and will discuss a predictive scheme that can guide the synthesis of new compositions with this structure type.
In the late 80's and early 90's, a significant number of oxides belonging to this family were synthesized and structurally characterized. In all cases, these oxides contained infinite chains of face-sharing octahedra and trigonal prisms, where the ratio of trigonal prisms to octahedra varied from a maximum of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 all the way to 0:1 for the idealized 2H-perovskite structure itself, which does not contain trigonal prisms. Using the known examples of this structure type, Darriet was able to formulate a general structural description that explained the existence of all the structural members belonging to the family of 2H-perovskite related oxides. Specifically, Darriet showed that this structure is generated from the AB stacking of two types of layers, an [A3O9] layer (tripled AO3 layer) and an [A3A′O6] layer.5,6 The latter derives from the A3O9 layer by substituting one A′ cation for 3 oxygen atoms. The specific stacking sequence of these two types of layers and the subsequent filling of the octahedral sites by B cations creates chains of face-sharing octahedra (BO6) and trigonal prisms (A′O6), where the ratio of trigonal prisms to octahedra is a function of the ratio of [A3O9] and [A3A′O6] layers. A general formula was derived, A3n + 3mA′nB3m + nO9m + 6n, where n/m is the ratio between the number of [A3A′O6] and [A3O9] layers (Fig. 1). As in the 2H-perovskite structure, the A cations separate the chains from one another. This general structural description accounts for all structures belonging to this family except for those that are incommensurate, where it is necessary to use a different formalism, A1 + x(A′xB1-x)O3 (0 ≤ x ≤ 0.5), that allows for non-integer ratios between n and m.7
:1 all the way to 0:1 for the idealized 2H-perovskite structure itself, which does not contain trigonal prisms. Using the known examples of this structure type, Darriet was able to formulate a general structural description that explained the existence of all the structural members belonging to the family of 2H-perovskite related oxides. Specifically, Darriet showed that this structure is generated from the AB stacking of two types of layers, an [A3O9] layer (tripled AO3 layer) and an [A3A′O6] layer.5,6 The latter derives from the A3O9 layer by substituting one A′ cation for 3 oxygen atoms. The specific stacking sequence of these two types of layers and the subsequent filling of the octahedral sites by B cations creates chains of face-sharing octahedra (BO6) and trigonal prisms (A′O6), where the ratio of trigonal prisms to octahedra is a function of the ratio of [A3O9] and [A3A′O6] layers. A general formula was derived, A3n + 3mA′nB3m + nO9m + 6n, where n/m is the ratio between the number of [A3A′O6] and [A3O9] layers (Fig. 1). As in the 2H-perovskite structure, the A cations separate the chains from one another. This general structural description accounts for all structures belonging to this family except for those that are incommensurate, where it is necessary to use a different formalism, A1 + x(A′xB1-x)O3 (0 ≤ x ≤ 0.5), that allows for non-integer ratios between n and m.7
 | ||
| Fig. 1 Representation of the formation of the end members of the A3m + 3nA′nB3m + nO9m + 6n family and some representative structures with both A3O9 and A3A′O6 layers. A′O6 trigonal prisms are shown in orange, BO6 octahedra are shown in blue, and A cations are shown in green. | ||
The A3n + 3mA′nB3m + nO9m + 6n formula is a powerful tool for predicting compositions that may result in the formation of specific members of this family. Over time, however, it has become clear that while the ratio of cations is predicted correctly, not every elemental composition that meets the correct ratio results in the desired product. If we use the basic ABO3 perovskite structure as an example, we find that it is not enough to balance the cationic charges with the anionic ones. Making the connection between chemical composition and crystal structure in solid-state materials is influenced by many factors including size, ionicity, coordination preferences of metals, as well as by synthetic parameters such as pressure and temperature. Size in many cases is a dominant force and we shall look more closely at it.
For trivalent ABO3 structures there exist two extreme cases. The first consists of A and B cations of roughly the same size and of a size suitable for coordination within an octahedral site and the second consists of A cations large enough to form close-packed layers with O2− and where the B cation is small enough to adopt an octahedral coordination environment. Oxides in the first group tend to form in basic sesquioxide structures, such as corundum or ilmenite, while oxides of the second group form AO3 close-packed layer structures containing linked BO6 octahedra, such as cubic or hexagonal perovskites. One predictive method, therefore, is to use radius ratio rules, where one can map out a “phase diagram” based on the relative A and B cation sizes.8,9 This method, although very simple, works exceedingly well and most ABO3 structure types will be clustered on such a diagram, enabling the chemist to make structure predictions based on size and charge balance. While not perfect, it is a very useful guide for roughly predicting what probably will or won't form in a certain structure type. We believe that this type of structure prediction may well work for this family and is developed at the end of the Highlight, following the discussion on the structures and associated properties of recently synthesized members of this family.
In this Highlight we will start by discussing the synthesis of 2H-perovskite related oxides and provide an up-to-date listing of different compositions belonging to this rather complex structural family that have been published since the paper by Stitzer et al. Following this, we will try to make some generalizations about the compositions that have been reported and illustrate how radius ratio rules can help us predict new compositions that might form in this structure type based on known compositions that do.
2. Syntheses
The interest in structures that have chains and that therefore may exhibit pseudo-one-dimensional properties has prompted many groups to pursue the synthesis of this class of oxides and to target new compositions. As this is a very flexible structure type, many elements in a variety of oxidation states can be incorporated into the chains. Early synthetic approaches relied on traditional solid-state synthesis; however, rapid advances in the syntheses of new 2H-perovskite related oxides came about when approaches were developed to grow these oxides as single crystals out of molten salts i.e. fluxes. Alkali metal carbonate fluxes were perhaps the most successful, however, other fluxes such as alkali metal and alkaline-earth metal hydroxides and halides have been used with almost equal success. The availability of single crystals immediately allowed for precise structural characterization and the investigation of physical properties using single crystals to determine their dependence on crystal orientation.The syntheses of these oxides are known to be very sensitive to the specific reaction conditions and, particularly when using elements that can take on multiple oxidation states, such as Rh or Ir, small variations in the synthesis conditions of powders can lead to significant deviations from the ideal structure (commensurate to incommensurate transition) and composition (oxygen content). Great care must be taken during the synthesis to control the oxygen partial pressure, heating rate, temperature, and time in order to prepare a unique sample composition and, consequently, sample structure. Primarily this applies to powder preparations; however, single crystal syntheses are not entirely exempt as heating rates, time, and flux to reactant ratios all play an important role in influencing the identity of the product that forms.
Flux growth uses a high temperature melt of inorganic compounds as the solvent from which crystals are grown.10–12 Typical fluxes consist of simple inorganic salts, such as metal halides, hydroxides, carbonates, and binary oxides. Of course, more complicated melts can also be used. To choose the “right” flux, one needs to consider the type of material one desires to crystallize and then to choose a melt that: is able to dissolve significant amounts of the starting materials used, that is low melting, that itself does not become incorporated into the product and that can be readily removed (dissolved) in order to isolate the crystals. There is not a single unique melt that one must use as typically a number of different fluxes will all result in crystals. However, for any given composition, some will work better than others. In a typical experiment the reagents are dissolved in the flux of choice and crystallization occurs as the solution becomes critically supersaturated. Supersaturation is usually achieved by changing the temperature, which initiates the nucleation and crystal growth process.
Single crystal growth is typically the preferred synthesis method as single crystals have the advantage that their structures can be readily determined; however, the preparation of polycrystalline samples is often necessary when large quantities of pure powders are needed for neutron diffraction studies to determine both the crystal and the magnetic structure.13 Nonetheless, as crystal growth techniques for these oxides continue to improve, the fabrication of large single crystals with a volume of 1 mm3, large enough for modern neutron diffraction facilities, can soon become a reality.
We will broadly differentiate between solid-state syntheses and flux growth, and in our discussion of the compositions and structures we will indicate the method of preparation (Table 1).
| Formula | Synthesisa | Structure Refinementb | Properties | Reference | |
|---|---|---|---|---|---|
| Magnetic | Optical | ||||
| m = 0, n = 1, x = 0.5 | |||||
| Ba3BaPtO6 [Ba4PtO6] | SS | PND | 60 | ||
| Ba3NaBiO6 | FG(Ba(OH)2/NaOH) | SC | 57 | ||
| Ba3NaIrO6 | FG(BaO/Ba2O2) | SC | 61 | ||
| Ba3NaNbO6 | FG(BaO/NaO0.52) | SC | 62 | ||
| Ba3NaRuO6 | FG(BaO/Ba2O2) | SC | 61 | ||
| Ba3NaSbO6 | FG(Ba(OH)2/NaOH) | SC | 14 | ||
| Ba3NaTaO6 | FG(Na2O) | SC | 62 | ||
| (Ba0.41Sr2.59)NaBiO6 | FG(Ba(OH)2/NaOH/Sr(OH)2) | SC | 18 | ||
| Ca3CaIrO6 [Ca4IrO6] | SS | PXRD | * | 63 | |
| Ca3CaPdO6 [Ca4PdO6] | SS | PXRD | 64 | ||
| Ca3CaPtO6 [Ca4PtO6] | FG(Na2CO3) | SC | 65 | ||
| Ca3(Ca0.5Cu0.5)PtO6 [Ca3.5Cu0.5PtO6] | FG | SC | 66 | ||
| Ca3(Ca0.1Cu0.9)RuO6 [Ca3.1Cu0.9RuO6] | SS | PND | * | 67 | |
| Ca3(Ca0.15Li0.85)IrO6 [Ca3.15Li0.85IrO6] | FG(K2CO3/KCl) | SC | * | 68 | |
| Ca3(Ca0.5Ni0.5)IrO6 [Ca3.5Ni0.5IrO6] | SS | PXRD | * | 69 | |
| Ca3(Ca0.75Ni0.25) IrO6 [Ca3.75Ni0.25IrO6] | FG(CaCl2/KCl/NaCl) | SC | * | 69 | |
| Ca3Co2O6 | FG(K2CO3), SS | PXRD, SC | * | 28,30 | |
| Cr-doped Ca3Co2O6 | SS | PXRD | * | 34 | |
| Ca3Co2-xFexO6 (x = 0, 0.1, 0.2, and 0.4) | SS | PND | * | 32 | |
| Ca3(Co1-xFex)2O6 (x = 0.1, 0.2 and 0.5) | SS | PXRD | * | 33 | |
| Ca3Co1 + yIr1-yO6 (y = 0.1, 0.3, 0.5, 0.7, 0.9) | SS | PXRD | * | 70 | |
| Ca3Co(Co0.25Mn0.75)O6 [Ca3Co1.25Mn0.75O6] | SS | PXRD | * | 27 | |
| Ca3Co(Co0.34Rh0.66)O6 [Ca3Co1.34Rh0.66O6] | FG(K2CO3) | SC | * | 26 | |
| Ca3Co1 + yRu1-yO6 (y = 0.1, 0.3, 0.5, 0.7, 0.9) | SS | PXRD | * | 70 | |
| Ca3CoIrO6 | SS | PXRD | * | 70 | |
| Ca3CoMnO6 | SS | PND, PXRD | * | 27 | |
| Ca3CoRhO6 | SS | PXRD | * | 71 | |
| Ca3CoRuO6 | SS | PXRD | * | 70 | |
| Ca3CuIrO6 | FG(CaF2/KF) | SC | 72 | ||
| Ca3CuMnO6 | SS | PND, PXRD | * | 27 | |
| Ca3CuRhO6 | FG(K2CO3) | SC | * | 26 | |
| Ca3FeRhO6 | FG(K2CO3), SS | PXRD | * | 26,71 | |
| Ca3LiOsO6 | FG(KCl/LiCl) | SC | * | 17 | |
| Ca3LiRuO6 | FG(K2CO3/KCl), SS | PXRD, SC | * | 13,68,73 | |
| Ca3LiSbO6 | SS | PXRD | 16 | ||
| Ca3MgIrO6 | FG(CaCl2/KCl/NaCl) | SC | 24 | ||
| Ca3NaIrO6 | FG(Na2CO3) | SC | 74 | ||
| Ca3NaRuO6 | FG(Na2CO3), SS | PXRD, SC | * | 13,73,74 | |
| Ca3NiMnO6 | SS | PND | * | 75,76 | |
| Ca3ZnCoO6 | SS | PND | * | 75 | |
| Ca3ZnIrO6 | FG(CaCl2/KCl/NaCl) | SC | 24 | ||
| Ca3ZnMnO6 | SS | PND | * | 75 | |
| (Ca2.7Sr0.3)Co2O6 | SS | PXRD | 31 | ||
| (La2.47Na0.53)NaRhO6 [La2.47Na1.53RhO6] | FG(NaOH) | SC | 21 | ||
| (La2Na)NaPtO6 [La2Na2PtO6] | FG(NaOH) | SC | 20 | ||
| (NaNd2)NaPtO6 [Nd2Na2PtO6] | FG(NaOH) | SC | 21 | ||
| (Na0.55Nd2.45)NaRhO6 [Nd2.45Na1.55RhO6] | FG(Na2CO3) | SC | 21 | ||
| (Na0.55Pr2.45)NaRhO6 [Pr2.45Na1.55RhO6] | FG(Na2CO3) | SC | 21 | ||
| Sr3CaRhO6 | SS | PXRD | * | 77 | |
| Sr3CdIrO6 | SS | PXRD | * | 77 | |
| Sr3CdPtO6 | FG(KOH) | SC | 78 | ||
| Sr3Co2O6 | SS | PXRD | * | 35 | |
| Sr3CoPtO6 | SS | PXRD | * | 79 | |
| Sr3(Co0.1In0.9)CoO6 [Sr3In0.9Co1.1O6] | SS | PXRD | * | 36 | |
| Sr3CuIrO6 | SS | PXRD | * | 80 | |
| Sr3Cu(Ir0.5Pt0.5)O6 | SS | PXRD | * | 81 | |
| Sr3CuRhO6 | SS | PXRD | * | 23 | |
| Sr3DyRhO6 | SS | PXRD | * | 83 | |
| Sr3ErCrO6 | SS | PXRD | * | 82 | |
| Sr3ErRhO6 | SS | PXRD | * | 83 | |
| Sr3EuRhO6 | SS | PXRD | * | 83 | |
| Sr3GdRhO6 | SS | PXRD | * | 84 | |
| Sr3HoCrO6 | SS | PXRD | * | 82 | |
| Sr3HoRhO6 | SS | PXRD | * | 83 | |
| Sr3InCrO6 | SS | PXRD | * | 82 | |
| Sr3InNiO6 | SS | PXRD | * | 85 | |
| Sr3InRhO6 | SS | PXRD | * | 86 | |
| Sr3LiBiO6 | FG(LiOH/Sr(OH)2) | SC | 57 | ||
| Sr3LiIrO6 | SS | PXRD | * | 87 | |
| Sr3LiNbO6 | FG(KOH/LiOH) | SC | * | 14 | |
| Sr3LiRhO6 | FG(KOH/LiOH) | SC | * | 88 | |
| Sr3LiRuO6 | SS | PXRD | * | 73 | |
| Sr3LiSbO6 | FG(LiOH/Sr(OH)2), SS | PXRD, SC | * | 14,16 | |
| Sr3LiTaO6 | FG(KOH/LiOH) | SC | * | 14 | |
| (Sr0.9La0.1)3NiPtO6 | SS | PXRD | * | 22 | |
| Sr3LuCrO6 | SS | PXRD | * | 82 | |
| Sr3LuNiO6 | SS | PXRD | * | 85 | |
| Sr3MgIrO6 | SS | PXRD | * | 89 | |
| Sr3MgPtO6 | SS | PXRD | * | 89 | |
| Sr3MgRhO6 | SS | PXRD | * | 89 | |
| Sr3NaBiO6 | FG(LiOH/Sr(OH)2) | SC | 57 | ||
| Sr3NaNbO6 | FG(NaOH/Sr(OH)2) | SC | * | 15 | |
| Sr3NaIrO6 | FG(Na2O2/SrO), SS | PXRD, SC | * | 87,90 | |
| Sr3NaRhO6 | FG(NaOH) | SC | * | 88 | |
| Sr3NaRuO6 | FG(Na2O2/SrO), SS | PXRD, SC | * | 73,90 | |
| Sr3NaSbO6 | SS | PND | 16 | ||
| Sr3NaTaO6 | FG(NaOH/Sr(OH)2) | SC | * | 15 | |
| Sr3NiIrO6 | SS | PXRD | * | 80 | |
| Sr3NiPtO6 | FG(K2CO3), SS | PXRD, SC | * | 91,92 | |
| Sr3NiRhO6 | SS | PXRD | * | 23 | |
| Sr3PbNiO6 | FG(NaCl), SS | SC | * | 93 | |
| Sr3(Pd0.25Sr0.75)PdO6 [Sr3.75Pd1.25O6] | SS | PXRD | 64 | ||
| Sr3ScCrO6 | SS | PXRD | * | 82 | |
| Sr3ScNiO6 | SS | PXRD | * | 85 | |
| Sr3ScRhO6 | SS | PXRD | * | 86 | |
| Sr3SmRhO6 | SS | PXRD | * | 83 | |
| Sr3SrIrO6 [Sr4IrO6] | SS | PND | 94 | ||
| Sr3SrPdO6 [Sr4PdO6] | SS | PXRD | 64 | ||
| Sr3SrPtO6 [Sr4PtO6] | SS | PXRD | 3 | ||
| Sr3SrRhO6 [Sr4RhO6] | SS | PXRD | * | 77 | |
| Sr4-yCayIrO6 (y = 0.5, 1, 2, 3, 4) | SS | PXRD | * | 87 | |
| Sr3(Sr0.67Na0.33)PtO6 [Sr3.67Na0.33PtO6] | FG(Na2O2/SrO2) | SC | 95 | ||
| Sr3TbRhO6 | SS | PXRD | * | 83 | |
| Sr3TmCrO6 | SS | PXRD | * | 82 | |
| Sr3TmNiO6 | SS | PXRD | * | 85 | |
| Sr3YCrO6 | SS | PXRD | * | 82 | |
| Sr3YRhO6 | SS | PXRD | * | 86 | |
| Sr3YbCrO6 | SS | PXRD | * | 82 | |
| Sr3YbNiO6 | SS | PXRD | * | 85,96 | |
| Sr3YbRhO6 | SS | PXRD | * | 83 | |
| Sr3ZnIrO6 | SS | PXRD | * | 80 | |
| Sr3ZnPtO6 | FG(KOH), SS | PXRD, SC | 25,78 | ||
| Sr3ZnRhO6 | SS | PXRD | * | 87,97,98 | |
| (Sr1-xNax)3NiPtO6 (x = 0.1, 0.2) | SS | PXRD | * | 22 | |
| m = 1, n = 1, x = 0.2 | |||||
|---|---|---|---|---|---|
| Ba6CuMn4O15 | SS | PND | * | 103 | |
| Ba6CuIr4O15 | SS | PXRD | 104 | ||
| Ba6Ni5O15 | FG(KOH) | SC | * | 105 | |
| Ba6ZnMn4O15 | SS | PND | * | 103 | |
| (Sr1-xBax)6Co5O15 (0 ≤ x ≤ 0.25) | SS | ED, HREM, PXRD | 106 | ||
| Sr6Co4.9Ni0.1O14.36 | FG(K2CO3) | SC | 38 | ||
| Sr6Co5O14.70 | FG(K2CO3) | SC | * | 38 | |
| Sr6Co5O15 | SS | PND | 37 | ||
| Sr6Rh5O15 | FG(K2CO3), SS | PXRD, SC | * | 39,107 | |
| m = 1, n = 3, x = 1/3 | |||||
|---|---|---|---|---|---|
| Ba4NaMn2O9 | FG(NaOH) | SC | 46 | ||
| Ba4Pt3O9 | FG(BaCl2) | SC | 110 | ||
| Ba12[BaxPt3-x]Pt6O27 (0 ≤ x ≤ 3) | SS | ED, PXRD | 111 | ||
| (Sr0.5Ca0.5)4Co3O9 | SS | ED, HREM, PXRD | 106 | ||
| (Sr3Ca)CoMn2O9 | SS | ED, HREM, PXRD | * | 48 | |
| (Sr3.3Ca0.7)CoRh2O9 | SS | ED, HREM, PXRD | * | 49 | |
| Sr4CoMn2O9 | SS | ED, HREM, PXRD | * | 47 | |
| Sr4CuIr2O9 | SS | HREM, PND | * | 112 | |
| Sr4CuMn2O9 | FG(K2CO3), SS | PND, SC | * | 43 | |
| Sr4Fe0.73Rh2.27O9 | FG(K2CO3) | SC | * | 50 | |
| Sr4LiMn2O9 | SS | PND, PXRD | * | 44 | |
| Sr4NiMn2O9 | FG(K2CO3) | SC | * | 45 | |
| Sr4Ni3O9 | FG(KOH) | SC | * | 113,114 | |
| Sr4ZnMn2O9 | SS | PND | * | 43 | |
| m = 2, n = 1, x = 0.125 | |||||
|---|---|---|---|---|---|
| Ba9Rh8O24 | FG(K2CO3) | SC | * | 115 | |
| m = 3, n = 1, x = 1/11 | |||||
|---|---|---|---|---|---|
| Ba12Rh9.25Ir1.75O33 | FG(K2CO3) | SC | * | 52 | |
| m = 8, n = 3, x = 0.1 | |||||
|---|---|---|---|---|---|
| Ba11Rh10O30 | FG(K2CO3) | SC | 53 | ||
| m = 23, n = 9, x = 3/29 | |||||
|---|---|---|---|---|---|
| Ba32Rh29O87 | FG(K2CO3) | SC | 53 | ||
| Incommensurate members A 1 + x (A′ x B 1-x )O 3 | |||||
|---|---|---|---|---|---|
| a For the Synthesis, FG = flux growth (of crystals) and SS = solid-state preparation (of polycrystalline material). b For the Structure Refinement, ED = electron diffraction, HREM = high-resolution electron microscopy, PND = powder neutron diffraction, PXRD = powder X-ray diffraction, and SC = single-crystal X-ray diffraction. | |||||
| Ba1.1170Ni0.1170Rh0.8830O3 | FG(K2CO3) | SC | 106,119 | ||
| Ba1.1605(Cu0.1605Rh0.8395)O3 | FG(K2CO3) | SC | 120 | ||
| Ba1.1695(Cu0.1695Rh0.8305)O3 | FG(K2CO3) | SC | 120 | ||
| Ba1.1667Pd0.1667Mn0.8333O3 | SS | ED, PXRD | * | 121 | |
| Ba1.2Mg0.2Mn0.8O3 | SS | PXRD | * | 122 | |
| Ba1.2Ni0.2Mn0.8O3 | SS | PXRD | * | 122 | |
| Ba1.2Pd0.2Mn0.8O3 | SS | ED, PXRD | * | 121 | |
| Ba1.2Zn0.2Ir0.8O3 | SS | PXRD | 104 | ||
| Ba1.2124Ni0.2124B0.7876O3 (B = 0.51Ni + 0.49 Pt) | FG(K2CO3) | SC | 123 | ||
| Ba1.25Pd0.25Mn0.75O3 | SS | ED, PXRD | * | 121 | |
| Sr1.266(Co0.266Mn0.734)O3 | SS | PND | * | 124 | |
| Ba1.2708(Cu0.2708Ir0.7292)O3 | FG(K2CO3) | SC | 125 | ||
| Sr1.280(Co0.280Mn0.720)O3 | SS | PND | * | 124 | |
| Sr1.2872NiO3 | FG(KOH) | SC | 126 | ||
| Sr1.31Co0.63Mn0.37O3 | SS | PND, PXRD | * | 127 | |
| Ba1.317(Cu0.39Pt0.61)O3 | FG(CuO) | SC | 128–130 | ||
| Sr1.32Mn0.83Cu0.17O3 | SS | ED, HREM, PXRD | 131 | ||
| Sr1.324(Cu0.3244Mn0.6756)O3 | SS | PXRD | 51 | ||
| Ba1.333Cu0.333Ir0.667O3 | SS | ED, HREM, PXRD | * | 112 | |
| Ba1.333Zn0.333Ir0.667O3 | SS | ED, HREM, PXRD | * | 112 | |
| Ca1.333Cu0.333Mn0.667O3 | SS | PXRD | * | 132 | |
| Sr1.333Cu0.333Mn0.667O3 | SS | PXRD | * | 132 | |
3. Members of the A3n + 3mA′nB3m + nO9m + 6n family
m = 0, n = 1
The largest number of compositions fall into the m = 0, n = 1 group, as it seems this structure type is the most flexible in terms of size and cation charges it can accommodate. The composition where m = 0, n = 1 is one end-member of the general family A3n + 3mA′nB3m + nO9m + 6n. Its structure consists of infinite chains of alternating trigonal prisms and octahedra occupied by the A′ and B cations, respectively, and where the A cations in square antiprismatic coordination environments function to separate the chains (Fig. 2). This composition contains the maximum number of trigonal prisms within the chains, i.e. 50%, such that the octahedra and trigonal prisms alternate in a 1:1 ratio. The other end-member of this general family where m = 1, n = 0 has no trigonal prisms in the infinite chains. All other members of this general family will possess less than 50% trigonal prisms in the infinite chains.
 | ||
| Fig. 2 Crystal structure of A3A′BO6. A′O6 trigonal prisms are shown in orange, BO6 octahedra are shown in blue, and A cations are shown in green. A3A′BO6 corresponds to the m = 0, n = 1 member of the A3m + 3nA′nB3m + nO9m + 6n family, and the x = 0.5 member of the A1 + x(A′xB1-x)O3 family. | ||
The basic composition of this member is A3A′BO6, where the A′ cation occupies the trigonal prismatic site and the B cation occupies the octahedral site. Assuming A is a divalent alkaline earth cation, then the possible oxidation state combinations for A′ and B are (I)/(V), (II)/(IV), (III)/(III), and (IV)/(II). It used to be standard that the A position was occupied by an alkaline-earth cation, however, more recently, mixed alkali, alkaline-earth and rare-earth cation combinations have been found and, as a result, other A′ and B oxidation state combinations, such as (I)/(IV), have recently been reported. The majority of the compositions discussed below were prepared as single crystals, however, some of the compositions were prepared as powders for use in neutron diffraction experiments.
Single crystals of Sr3LiNbO6, Sr3NaNbO6, Sr3LiTaO6, and Sr3NaTaO614,15 were prepared from high-temperature solutions. Nb2O5 or Ta2O5 was dissolved at 700 °C in a flux composed of Sr(OH)2·8H2O and either LiOH·H2O/KOH or NaOH. In these structures, the trigonal prismatic sites are fully occupied by Li+ or Na+, and the octahedral sites are fully occupied by Nb5+ or Ta5+. For all four compounds, the crystals were pale brown when grown in open silver crucibles; however, Sr3NaNbO6 and Sr3NaTaO6 could also be grown as green crystals in sealed silver tubes. X-ray diffraction analysis (powder and single-crystal) and elemental analysisvia energy-dispersive spectroscopy showed the same composition for both colors of crystals. The difference in color of the crystals has been attributed to crystal defects and morphological differences owing to the different synthetic conditions. UV-visible absorbance spectra on all four compounds are consistent with the colors of the powders (ground crystals). Additionally, both Sr3NaNbO6 and Sr3NaTaO6 luminesce with a bluish-violet emission upon UV excitation at 250 nm (Fig. 3).
 | ||
| Fig. 3 Optical photograph of powdered brown crystals of Sr3NaTaO6 under ambient light (left) and under UV irradiation at 250 nm (right). Reprinted from Ref. 15 with permission. Copyright 2008, American Chemical Society. | ||
Other (I)/(V) compositions, Ca3LiSbO6 and Sr3LiSbO6,16 were prepared as polycrystalline powders from ACO3 (A = Ca, Sr), Li2CO3, and Sb2O3 heated initially at 600 °C. The samples were re-heated at increasing temperatures until pure samples were obtained, with final temperatures of 800 °C for Ca3LiSbO6 and 950 °C for Sr3LiSbO6. In these phases, the A site is occupied by Ca2+ or Sr2+, the trigonal prismatic site is occupied by Li+, and the octahedral site is occupied by Sb5+.
Pale brown crystals of Ba3NaSbO614 were prepared from Sb2O3 in a molten flux consisting of Ba(OH)2·8H2O and NaOH at 700 °C. Although this compound had been synthesized previously as a polycrystalline powder,16 only the lattice parameters had been reported due to the instability of the material. Therefore, the single-crystal X-ray structure data collected here represented the first full structure refinement, which revealed the presence of Na+ on the trigonal prismatic site and Sb5+ on the octahedral site.
Shiny black hexagonal crystals of Ca3LiOsO617 (Fig. 4) were prepared at 750 °C from Ca3OsO6 in a flux of LiCl and KCl. This structure contains Li+ in the trigonal prismatic sites, as well as Os5+ in the octahedral sites. Ca3LiOsO6 exhibits an antiferromagnetic ordered state with a transition temperature of 117 K. Additionally, specific heat measurements on this compound have confirmed the temperature of the antiferromagnetic transition.
 | ||
| Fig. 4 Optical photograph of shiny black hexagonal crystals of Ca3LiOsO6. Reprinted from Ref. 17 with permission. Copyright 2010, American Chemical Society. | ||
(Sr2.59Ba0.41)NaBiO618 was prepared as single crystals at 600 °C from Bi2O3 in a molten flux of Sr(OH)2, Ba(OH)2, and NaOH. In this structure, the trigonal prismatic site contains Na+ and the octahedral site contains Bi5+. (Sr2.59Ba0.41)NaBiO6 was the first example of a 2H-perovskite related oxide to exhibit mixed divalent cations (Sr2+ and Ba2+) on the A site.
Crystals of Ca3.15Li0.85IrO619 having mixed A′ site occupancy were prepared from CaCO3, LiOH·H2O, and Ir metal in a molten mixture of K2CO3 and KCl at 1050 °C. In Ca3.15Li0.85IrO6, the trigonal prismatic site is occupied by 85% Li+ and 15% Ca2+. To achieve charge balance, the octahedral site must be filled by a combination of Ir4+ and Ir5+. Magnetic susceptibility measurements reveal no magnetic ordering down to 2 K, and indicate only simple paramagnetic behavior consistent with the presence of Ir4+.
Yellow prismatic crystals of (NaLn2)NaPtO6 (Ln = La, Nd)20,21 (Fig. 5) were prepared from Ln2O3 and (NH4)2PtCl6 in a NaOH melt at 700 °C. In these compounds, the trigonal prismatic sites are occupied by Na+ and the octahedral sites by Pt4+. Interestingly, (NaLa2)NaPtO6 was the first 2H-perovskite related oxide to exhibit occupation of the A site by a rare-earth (which occupies the site in combination with Na+ cations); the Nd analog was reported shortly thereafter. Magnetic susceptibility measurements on (NaNd2)NaPtO6 display only paramagnetic behavior due to the non-interacting magnetic moments of the Nd3+ cations. These two compounds remain the only known examples of an m = 0, n = 1 member that has the unusual (I)/(IV) cation charge combination.
 | ||
| Fig. 5 Scanning electron micrograph of a prismatic crystal of (NaLa2)NaPtO6. Reprinted from Ref. 20 with permission. Copyright 2003, American Chemical Society. | ||
Single crystals of (Ln3-xNax)NaRhO6 (Ln = La, x = 0.53; Ln = Pr, Nd, x = 0.55)21 phases were grown from fluxes. La2.47Na1.53RhO6 was prepared from La2O3 and Rh2O3 in molten NaOH at 700 °C, while Pr2.45Na1.55RhO6 and Nd2.45Na1.55RhO6 were prepared from Ln2O3 and Rh metal in a Na2CO3 flux at 1050 °C. The crystals of the La compound were brown-green, and the Pr and Nd analogs were both black. In these phases, the square antiprismatic A site is occupied by a mixture of Ln3+ and Na+ and the trigonal prismatic site is fully occupied by Na+. The octahedral site is proposed to be filled primarily by Rh3+ and a very small amount of Rh4+, where this charge distribution is hypothesized solely on the basis of refined site occupancies from the single-crystal X-ray structure solution.
Other compositions that have mixed A site occupancies include (Sr1-xAx)3NiPtO6 (A = La, x = 0.1; A = Na, x = 0.1, 0.2)22 which were prepared as polycrystalline powders via a high-temperature solid-state route. SrCO3, La2O3 or Na2CO3, NiO, and PtO2 were heated in air at 1000 °C for 10 days with intermittent grindings. In these materials, the A site is occupied by a mixture of Sr2+ and either La3+ or Na+, the trigonal prismatic sites are occupied by Ni, and the octahedral sites are occupied by Pt. X-ray photoelectron spectroscopy on the parent compound Sr3NiPtO6, as well as these mixed phases, suggests that the substitution of Sr2+ by La3+ or Na+ can be used to subtly change the oxidation states of both Ni (Ni2+, Ni3+, and Ni4+) and Pt (Pt2+, Pt3+, Pt4+). Magnetic susceptibility measurements indicate a spin-liquid-like behavior for these compounds where Ni is in the high-spin state and Pt is in the low-spin state.
There are a large number of (II)/(IV) combinations including both Sr3NiRhO6 and Sr3CuRhO6,23 which were prepared as polycrystalline powders by standard high-temperature solid-state syntheses. SrCO3, NiO or CuO, and Rh metal were pressed into a pellet and heated at 1000 °C in air, with intermittent grindings, for a total of 15 days. Both commensurate and incommensurate phases (Sr3MRhO6 + δ) (M = Ni, Cu) could be synthesized by this technique. In order to minimize the formation of the incommensurate phase, the sample could not be heated for long periods of time because the transition to the incommensurate phase begins as soon as the commensurate phase has formed. Thermogravimetric analysis was used to confirm the stoichiometric oxygen content of the commensurate phases. In both of these structures, Rh4+ occupies the octahedral site, and Ni2+ or Cu2+ occupies the trigonal prismatic site. Because Cu prefers a square planar coordination, the Cu2+ cation moves toward the face of the trigonal prism, to achieve a pseudo-square planar coordination, whereas Ni2+ is located in the center of the trigonal prism. The magnetic susceptibility of Sr3NiRhO6 shows a sudden drop at 30 K, indicating antiferromagnetic correlations, whereas Sr3CuRhO6 exhibits ferromagnetic-type ordering below 10 K.
Black single crystals of Ca3.34Mg0.66IrO6 and Ca3.50Zn0.50IrO624 were prepared from Ca(OH)2, MgO or ZnO, and Ir metal at 925 °C from a eutectic flux consisting of CaCl2, KCl, and NaCl. In both compounds, the octahedral sites are fully occupied by Ir4+, while the trigonal prismatic sites exhibit mixed occupancy by Ca2+ and either Mg2+ or Zn2+, respectively. Ca3.50Zn0.50IrO6 is a unique structure in that Zn adopts the exceedingly rare trigonal prismatic geometry25 as opposed to the much more common tetrahedral geometry.
Black single crystals of Ca3Co1.34Rh0.66O6 and Ca3CuRhO626 (Fig. 6) were prepared from CaCO3, Co3O4 or CuO, and Rh metal with a K2CO3 flux at 1050 °C. In Ca3Co1.34Rh0.66O6, the trigonal prismatic site is fully occupied by Co, while the octahedral site is occupied by a mixture of Co and Rh (34% and 66%). For Ca3CuRhO6, the Cu2+ cation is again shifted toward the face of a trigonal prism, allowing it to achieve a pseudo-square planar coordination, while the Rh4+ fully occupies the octahedral site. Magnetic susceptibility measurements showed Ca3CuRhO6 to be a soft ferromagnet with a lower than expected magnetic moment. A similar magnetic study indicated the presence of ferromagnetic correlations in Ca3Co1.34Rh0.66O6, but it was not possible to assign the oxidation states of Co and Rh on the basis of the effective magnetic moment.
 | ||
| Fig. 6 Scanning electron micrograph of a hexagonal crystal of Ca3CuRhO6. Reprinted from Ref. 26 with permission. Copyright 2003, Elsevier. | ||
Polycrystalline powders of Ca3CoMnO6, Ca3Co1.25Mn0.75O6, and Ca3Cu0.95Mn1.05O627 were prepared via a high-temperature solid-state route. CaCO3, MnO2, and either CoO or CuO were mixed and pressed into pellets, heated in air at 950 °C for 30 h and then at 1130 °C (Cu) or 1200 °C (Co) for 24 h, before a final anneal in O2 at 1000 °C for 4 h. In Ca3CoMnO6, the trigonal prismatic site is occupied by high-spin Co2+ and the octahedral site is occupied by Mn4+. For Ca3Co1.25Mn0.75O6, a mixture of high-spin Co2+ and high-spin Co3+ is located in the trigonal prism, while low-spin Co3+ and Mn4+ are in the octahedron. Ca3Cu0.95Mn1.05O6 is a rarity in the A3A′BO6 family in that it adopts the low symmetry triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group, rather than the more common hexagonal R
space group, rather than the more common hexagonal R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c space group. The result is that there are two A′ sites and two B sites instead of the usual one and one. Of the two trigonal prismatic sites, one is occupied fully by Cu2+, and the other is occupied by 90% Cu2+ and 10% Mn4+. The shift of the Cu atom toward the face of the trigonal prism is reduced in the site with mixed Mn occupation. Both octahedral sites are occupied by Mn, which is predominantly Mn4+, but some lower valent Mn must also be present to balance the charge on the A′ site. These oxidation states and spin states have been confirmed by magnetic susceptibility measurements. All three compounds order antiferromagnetically with transition temperatures of 18 K (Ca3Co1.25Mn0.75O6), 13 K (Ca3CoMnO6), and 6 K (Ca3Cu0.95Mn1.05O6).
c space group. The result is that there are two A′ sites and two B sites instead of the usual one and one. Of the two trigonal prismatic sites, one is occupied fully by Cu2+, and the other is occupied by 90% Cu2+ and 10% Mn4+. The shift of the Cu atom toward the face of the trigonal prism is reduced in the site with mixed Mn occupation. Both octahedral sites are occupied by Mn, which is predominantly Mn4+, but some lower valent Mn must also be present to balance the charge on the A′ site. These oxidation states and spin states have been confirmed by magnetic susceptibility measurements. All three compounds order antiferromagnetically with transition temperatures of 18 K (Ca3Co1.25Mn0.75O6), 13 K (Ca3CoMnO6), and 6 K (Ca3Cu0.95Mn1.05O6).
Ca3Co2O628–30 is perhaps the most studied composition, where it is believed that Co+3 occupies both the trigonal prismatic and octahedral sites. Many doped phases have been prepared and reported and examples include: 1) doping on the A site, for example, Sr2+ for Ca2+,31 and 2) doping on the A′ and/or B sites with other transition metals, for example, Fe32,33 or Cr34 in place of Co. These have typically been prepared as polycrystalline samples by standard high-temperature solid-state reactions.
A polycrystalline sample of Sr3Co2O635 was prepared from Co powder and SrO2 heated at 1400 °C under 6 GPa in a belt-type high-pressure apparatus. The structure was solved via a Rietveld refinement of synchrotron X-ray diffraction data, and thermogravimetric analysis confirmed the oxygen content. In this compound, both the octahedral and trigonal prismatic sites are occupied by Co, which is presumed to be Co3+. Magnetic susceptibility measurements reveal complex spin-chain magnetism in Sr3Co2O6 similar to what has been observed for Ca3Co2O6.
Sr3In0.9Co1.1O636 was prepared in polycrystalline form from SrCO3, In2O3, and Co3O4 heated in air at 1000 °C for 5 days with intermittent grindings. According to neutron diffraction, this structure possesses octahedral sites fully occupied by low-spin Co3+, and trigonal prismatic sites filled by a 9:1 ratio of In3+ to high-spin Co3+. A magnetic study of Sr3In0.9Co1.1O6 showed no magnetic ordering down to 1.5 K, instead indicating simple paramagnetic behavior due to the isolated high-spin Co3+ cations.
Clearly, a large variety of cations can occupy the different sites in this end member of a very complex structural family. By comparison, far fewer compositions are known for other m and n values, as discussed below, apparently because these compositions have tighter tolerance factors (size and charge) and fewer elemental compositions exist that can fulfill these stringent requirements.
m = 1, n = 1
The m = 1, n = 1 structure is built up from equal numbers of alternating A3O9 and A3A′O6 layers. An early example of this structure, which has four octahedra followed by one trigonal prism in the chain (Fig. 7), is Sr6Co5O15, which was studied by powder neutron diffraction in the 1990s.37 Attempts to grow single crystals of this composition from fluxes have resulted in oxygen deficient compounds. Single crystals of Sr6Co5O14.70 (Fig. 8) and Sr6Co4.9Ni0.1O14.3638 were prepared using a K2CO3 flux. Sr6Co5O14.70 was synthesized by heating SrCO3, Co2O3, and Cr2O3, with the atomic ratio Sr:Co:Cr = 6:5:1, in 20 g of K2CO3 at 920 °C. The Cr was not incorporated into the structure. Sr6Co4.9Ni0.1O14.36 was synthesized by heating SrCO3, Co2O3, and NiO with the atomic ratio Sr:Co:Ni = 6:5:1 by the same heating procedure. The structures were determined by single crystal X-ray diffractionvia both the traditional method as well as by using the 4-D superspace group approach.6Sr6Co5O14.70 exhibits antiferromagnetic ordering at about 25 K. A large magnetic anisotropy was observed in this compound, with a much larger magnetic susceptibility along the c axis vs. in the ab plane, consistent with what others have observed for this structural family; for example, Sr6Rh5O1539 exhibits an even larger magnetic anisotropy.
 | ||
| Fig. 7 Crystal structure of A6A′B4O15. A′O6 trigonal prisms are shown in orange, BO6 octahedra are shown in blue, and A cations are shown in green. A6A′B4O15 corresponds to the m = 1, n = 1 member of the A3m + 3nA′nB3m + nO9m + 6n family, and the x = 0.2 member of the A1 + x(A′xB1-x)O3 family. | ||
 | ||
| Fig. 8 Optical photograph of a black hexagonal prismatic crystal of Sr6Co5O14.70. Reprinted from Ref. 38 with permission. Copyright 2006, American Chemical Society. | ||
m = 1, n = 2
Compositions belonging to the m = 1, n = 2 structure have a 3 OH–1 TP–2 OH–1 TP (OH = octahedron; TP = trigonal prism) repeat sequence (Fig. 9) and include Sr9A2Mn5O21 (A = Co, Ni),40,41 which were synthesized by traditional solid-state syntheses. Sr9Ni2Mn5O21 was synthesized by heating stoichiometric amounts of SrCO3, MnCO3, and NiO in air at 1050 °C for 3 days. Sr9Co2Mn5O21 was synthesized by heating stoichiometric amounts of SrCO3, MnCO3, and Co3O4 in air at 1200 °C for 7 days. The compounds were characterized using SAED and HREM, and subsequently the crystal structures were solved by Rietveld refinement. The trigonal prismatic sites are occupied by Co2+ and Ni2+, while Mn4+ cations occupy the octahedral sites. Magnetic susceptibility measurements of Sr9Co2Mn5O21 did not indicate the presence of any long-range magnetic order. | ||
| Fig. 9 Crystal structure of A9A′2B5O21. A′O6 trigonal prisms are shown in orange, BO6 octahedra are shown in blue, and A cations are shown in green. A9A′2B5O21 corresponds to the m = 1, n = 2 member of the A3m + 3nA′nB3m + nO9m + 6n family, and the x = 2/7 member of the A1 + x(A′xB1-x)O3 family. | ||
m = 1, n = 3
Compositions belonging to the m = 1, n = 3 structure have a 2 OH–1 TP repeat sequence (Fig. 10) and are thus the closest to the m = 0, n = 1 members which have a 1 OH–1 TP repeat sequence. Perhaps this similarity is the reason why a relatively large number of examples of the m = 1, n = 3 phase exist. Single crystals of Sr4CuMn2O942 were synthesized from SrCO3, Mn2O3 and CuO in a K2CO3 flux at 927 °C. The related compositions, Sr4MMn2O9 (M = Cu, Zn)43 were synthesized by a solid-state route. Commensurate powders of Sr4CuMn2O9 were prepared by heating starting materials with a ratio Sr:Cu:Mn of 4:1.04:1.92, at 1200 °C in air for 6 days with intermittent grinding. The exact composition of the products was refined to be Sr4Cu1.01(1)Mn1.99(1)O9. The stoichiometric mixture of starting materials of SrCO3, CuO and MnO2 leads to an incommensurate structure. For the synthesis of the Zn analogue, a stoichiometric mixture of SrCO3, ZnO and MnO2 was intimately ground, calcined at 800 °C, pressed into pellets, and then heated at 1200 °C for 6 days with intermittent grindings. The structures were solved using powder neutron diffraction data, which indicated that the Cu2+ cations in the trigonal prismatic sites of Sr4CuMn2O9 are displaced toward the faces, instead of occupying the center of the trigonal prisms. In Sr4ZnMn2O9, there is evidence that a small amount of Mn is present in the trigonal prismatic site. Magnetic susceptibility measurements indicate that short-range antiferromagnetic interactions are present below 120 K.
 | ||
| Fig. 10 Crystal structure of A4A′B2O9. A′O6 trigonal prisms are shown in orange, BO6 octahedra are shown in blue, and A cations are shown in green. A4A′B2O9 corresponds to the m = 1, n = 3 member of the A3m + 3nA′nB3m + nO9m + 6n family, and the x = 1/3 member of the A1 + x(A′xB1-x)O3 family. | ||
Another isostructural compound, Sr4LiMn2O9,44 whose composition is reported as Sr4Li0.89Mn2.11O9 based on a combination of powder X-ray and powder neutron Rietveld refinements, was synthesized by microwave heating. For the preparation of this compound, LiMn2O4 was first synthesized by heating MnO2 and Li2CO3 in air at 800 °C. Then LiMn2O4 was allowed to react with SrCO3 at 950–1000 °C in a microwave furnace. There are two crystallographically distinct chains in the structure. In one of the two chains, Li exclusively occupies the trigonal prismatic sites and Mn exclusively occupies the octahedral sites, while in the other chain, Li and Mn are disordered over both the trigonal prismatic and the octahedral sites. The temperature dependent magnetic susceptibility data yielded an effective magnetic moment of 5.393 μB, close to the theoretical value of 5.192 μB based on 31.8% Mn5+ as required by the formula Sr4Li0.89Mn2.11O9.
Single crystals of Sr4NiMn2O945 were grown out of a K2CO3 flux at 1100 °C. In this compound, Mn occupies the octahedral sites and Ni the trigonal prismatic sites. Interestingly, the Ni2+ cations are disordered in the trigonal prisms, with roughly 20% occupying the center and roughly 80% displaced toward the faces of the trigonal prisms. Magnetic measurements demonstrate that this compound orders antiferromagnetically below 3 K. The high temperature susceptibility data are in good agreement with Mn4+ occupying the octahedral sites and 20% Ni2+ (S = 1) being located in the center of trigonal prisms, and 80% Ni2+(S = 0) located in the faces of trigonal prisms.
Single crystals of another Mn-containing m = 1, n = 3 phase, Ba4Mn2NaO9,46 were synthesized by electrosynthesis in molten NaOH. The mixture of NaOH, Ba(OH)2⋅3H2O, V2O5, Bi2O3 and MnO2 was heated at 800 °C with a constant applied potential of 2.2 V for 24 h. In this structure determination, the Na+ cations occupy the trigonal prismatic sites in the chain while Mn is again in the octahedral coordination sites. Based on the overall composition, the oxidation state of Mn is +4.5. However, there is no evidence for distinct Mn(IV) and Mn(V) sites in this crystal structure.
It has been possible to prepare members of this structure type with more than one alkaline-earth cation occupying the A site. Sr4-xCaxMn2CoO9 (x = 0, 1)47,48 were prepared via solid-state syntheses. Sr4Mn2CoO9 was synthesized by heating SrCO3, Co3O4 and MnO2 in air at 1275 °C for 5 days, while Sr3CaMn2CoO9 was synthesized by heating SrCO3, CaCO3, Co3O4 and MnO2 in air at 1300 °C for 7 days. The structures were characterized by SAED and HREM, and subsequently solved by Rietveld refinement of power X-ray diffraction data. Magnetic susceptibility data were collected for both compounds, where the large negative Weiss constant, −91 K, for Sr4Mn2CoO9 indicates that the magnetic interactions are predominantly antiferromagnetic. It was suggested that Sr4Mn2CoO9 was partially magnetically ordered below 9.5 K,47 while Sr3CaMn2CoO9 is reported to order antiferromagnetically at 13.5 K.48
Sr3.3Ca0.7CoRh2O949 has a similar mix of alkaline earth cations on the A site. The composition was synthesized by a solid-state route. The starting materials SrCO3, CaCO3, Co3O4 and Rh2O3 were initially heated in air at 900 °C for 11 h to decompose the carbonates, and then heated again at 1050 °C for 6 days with intermittent grindings. The structure was characterized by SAED and HREM, and subsequently solved by Rietveld refinement of power X-ray diffraction data. This compound features Co and Rh disordered over both the trigonal prismatic and octahedral sites. It was suggested that the drop of magnetic susceptibility at about 8.2 K can be interpreted as the transition into a partially ordered state.
An example of a m = 1, n = 3 member that has mixed metal occupancy of the trigonal prismatic site is Sr4Fe0.73Rh2.27O9.50 The single crystals (Fig. 11) were synthesized from SrCl2·6H2O, Fe2O3, and Rh out of a K2CO3 flux at 1050 °C. The trigonal prismatic sites are occupied by a mixture of Fe and Rh, while only Rh occupies the face-sharing octahedral sites. This compound orders antiferromagnetically below 8 K.
 | ||
| Fig. 11 Scanning electron micrograph of a columnar crystal of Sr4Fe0.72Rh2.27O9. Reprinted from Ref. 50 with permission. Copyright 2004, Elsevier. | ||
m = 1, n = 4
Compositions belonging to the m = 1, n = 4 structure have a complex and long repeat pattern of 1 TP-2 OH-1 TP-2 OH-1 TP-2 OH-1 TP-1 OH and include (Sr0.5Ca0.5)15Mn7Co4O33,48 which was synthesized by a solid-state method. The starting materials, SrCO3, CaCO3, Co3O4 and MnO2 were heated in air at 1450 °C for 5 days. (Sr0.5Ca0.5)15Mn7Co4O33 was structurally characterized by SAED and HRTEM. The structure was confirmed by Rietveld refinement of powder X-ray diffraction data. The compound orders antiferromagnetically below 16.5 K.m = 2, n = 3
An oxide having the m = 2, n = 3 structure, which has a repeat sequence of 1 TP–3 OH (Fig. 12), is (Sr0.75Ba0.25)5NiMn3O12,41 which was synthesized from a solid-state route, by heating stoichiometric amounts of SrCO3, BaCO3, MnCO3 and NiO in air at 1200 °C for 3 days. The structure was characterized by SAED, HREM and refined using powder X-ray diffraction data. This is another interesting example where the A site is occupied by two different cations, Sr2+ and Ba2+. The Ni cation occupies the trigonal prismatic site, while Mn is in the octahedral sites. | ||
| Fig. 12 Crystal structure of A5A′B3O12. A′O6 trigonal prisms are shown in orange, BO6 octahedra are shown in blue, and A cations are shown in green. A5A′B3O12 corresponds to the m = 2, n = 3 member of the A3m + 3nA′nB3m + nO9m + 6n family, and the x = 0.25 member of the A1 + x(A′xB1-x)O3 family. | ||
m = 5, n = 9
A related repeat sequence is found in the m = 5, n = 9 member Sr14Cu3Mn8O33,51 consisting of 3 OH-1 TP-2 OH-1 TP-3 OH. For the synthesis of single crystals, a ground mixture of SrCO3, Mn2O3, and CuO was heated in a K2CO3 flux at 1200 °C. The structure was solved by the (3 + 1)D superspace group approach. In this structure Cu occupies the trigonal prismatic site and, as seen before, the Cu cations are displaced toward the face, instead of occupying the center of the trigonal prism. All the octahedral sites are occupied by Mn.m = 3, n = 1
Structures with longer octahedral repeats include the m = 3, n = 1 members which have a repeat of 10 octahedra followed by one trigonal prism (Fig. 13). Crystals of Ba12Rh9.25Ir1.75O3352 were synthesized from BaCO3, Rh and Ir in a K2CO3 flux at 1150 °C. In Ba12Rh9.25Ir1.75O33, all the octahedral and trigonal prismatic sites in the chains have mixed Rh/Ir occupancy and some of the polyhedra exhibit significant distortion. Interestingly, an analysis of the polyhedral height shows that the polyhedra close to the trigonal prismatic site along with the trigonal prismatic site are the tallest ranging from 2.6–2.95 Å. The polyhedra in the middle of the sequence of octahedra have roughly the same height of, on average, 2.512 Å, versus an ideal value of 2.366 Å. As a result, the trigonal prismatic site is only 2.958 Å versus an ideal value of 4.73 Å. The magnetic susceptibility plot of this composition is featureless, showing no sign of long-range magnetic order down to 2 K.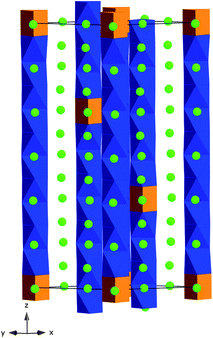 | ||
| Fig. 13 Crystal structure of A12A′B10O33. A′O6 trigonal prisms are shown in orange, BO6 octahedra are shown in blue, and A cations are shown in green. A12A′B10O33 corresponds to the m = 3, n = 1 member of the A3m + 3nA′nB3m + nO9m + 6n family, and the x = 1/11 member of the A1 + x(A′xB1-x)O3 family. | ||
m = 8, n = 3
A structure with an only slightly shorter octahedral repeat sequence is the m = 8, n = 3 member, which has a repeat sequence of nine octahedra followed by one trigonal prism (Fig. 14). An example is Ba11Rh10O30,53 which was obtained as single crystals from BaCO3 and Rh in a K2CO3 flux at 1050 °C. The structure was solved with the (3 + 1)D superspace formalism approach and contains Rh cations in both the octahedral and the trigonal prismatic sites. An investigation of the Rh-Rh separation along the chain revealed that the distance is largest between Rh atoms in the trigonal prisms and the adjacent octahedra, ∼2.75 Å, while those between adjacent octahedra in the middle of the chain are the shortest at ∼2.47 Å. | ||
| Fig. 14 Crystal structure of A11A′B9O30. A′O6 trigonal prisms are shown in orange, BO6 octahedra are shown in blue, and A cations are shown in green. A11A′B9O30 corresponds to the m = 8, n = 3 member of the A3m + 3nA′nB3m + nO9m + 6n family, and the x = 0.1 member of the A1 + x(A′xB1-x)O3 family. | ||
m = 23, n = 9
The longest repeat sequence reported to date is found in the m = 23, n = 9 member, represented by Ba32Rh29O87,53 which has a repeat sequence of 9 OH-1 TP-9 OH-1TP-8 OH-1TP (Fig. 15). Crystals were synthesized from BaCO3 and Rh in a K2CO3 flux at 1050 °C, and the structure was solved using the (3 + 1)D superspace formalism approach. Rh occupies all the polyhedral sites in the chain and the trend of Rh–Rh distances mimics that observed for Ba11Rh10O30.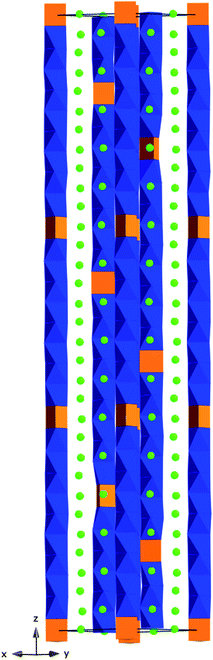 | ||
| Fig. 15 Crystal structure of A32A′3B26O87. A′O6 trigonal prisms are shown in orange, BO6 octahedra are shown in blue, and A cations are shown in green. A32A′3B26O87 corresponds to the m = 23, n = 9 member of the A3m + 3nA′nB3m + nO9m + 6n family, and the x = 3/29 member of the A1 + x(A′xB1-x)O3 family. | ||
One can surmise that other compositions can be prepared that have even longer repeats and, in fact, if one considers the incommensurate compositions, then the repeat sequence of octahedra and trigonal prisms is non-repeating and is “infinite” in length.
4. Predictions
A very large number of compositions belonging to the A3n+3mA′nB3m+nO9m+6n family have been prepared and structurally characterized. To enable further progress in synthesizing new oxides belonging to this family it becomes necessary to understand whythese specific compositions form in this structure type. Armed with such an understanding, it will be possible to predict new compositions that also belong to this family. Historically, many approaches have been used for structural predictions and most have incorporated some of the many interactions that factor into the stability of a structure, such as lattice energies, crystal-field stabilization energies, geometric packing effects, ionicity and bond directionality.54 A simple but very effective correlation using radius ratios was introduced by Roth, further elaborated by Muller and Roy, and later modified by Giaquinta to predict structure types for diverse ABO3 compositions.8,9,55 This simple approach works exceedingly well and achieves clustering of most ABO3 structure types on such plots, which allows the user to both rationalize why a certain composition forms in a given structure type as well as predict which structure type certain compositions should form in. We therefore decided to see if this radius ratio approach, modified slightly to accommodate the diverse A3n+3mA′nB3m+nO9m+6n structures and compositions, would also work for the 2H-perovskite related family of oxides.It is important to keep in mind that the goal of the radius ratio-based structural analysis that follows is not to predict the probable structure for any given elemental composition, but rather to ascertain, based on the 2H-perovskite related oxides that have been synthesized already, if a given combination of cations can feasibly take on a 2H-perovskite related structure.
In order to analyze the many members of the 2H-perovskite related oxide family for correlations with respect to structure, the reported structures were tabulated and only compositions with concrete site occupancies and stoichiometries were included. Using the wealth of structures reported in the literature, a radius ratio plot, depicting the approximate phase space of the 2H-perovskite related structures, is shown in Fig. 16. Using Shannon's radii,56 the A′ and B radii of the cations in 2H-perovskite related oxides were plotted against each other to generate a plot that can be used to empirically extrapolate what phases might form for a given set of A′ and B cations.
 | ||
| Fig. 16 Radius ratio plot of rA′vs. rB for the A3m + 3nA′nB3m + nO9m + 6n family of oxides. The region corresponding to the m = 1, n = 0 members is shaded in blue, while the region corresponding to all other m and n values is shaded in orange. Data points are omitted for clarity and compositions at the limits are labeled. Data for ABO3 2H-perovskites are also included and are plotted assuming rA′ = rB for comparison. The m = 0, n = 1 compositions can form for a wide range of radius ratios, while the compositions corresponding to other m and n values fall into a much more restricted region. | ||
A few basic boundaries limit the range of existence for the m = 0, n = 1 members of 2H-perovskite related structures. First, the six-coordinate A′ cation must be smaller than the eight-coordinate A cation. Often, the largest A′ cation that is observed is A itself; Sr4PtO63 is an example of this. Second, it is apparent that the trigonal prismatic site is larger than the octahedral site; hence, the A′ cation is expected to be larger than the B cation for any composition. Some compositions seem to violate this, for example when both A′ and B are predicted to have the same ionic radius as in Sr3LiBiO657 (Li+ = 0.76 Å, Bi5+ = 0.76 Å), or when the same cation occupies both the octahedral and the trigonal prismatic sites, such as in A3Co2O6 (A = Sr, Ca). However, evidence exists to support spin ordering in the A3Co2O6 compositions with high-spin Co3+ in the trigonal prism and low-spin Co3+ in the octahedra.58 For the case of Sr3LiBiO6, it should be noted the Shannon radii do not distinguish between octahedral and the larger trigonal prismatic coordination environments. Since the trigonal prismatic coordination environment is a rarity in oxides, no data exists specifically for cation radii in trigonal prismatic coordination environments in oxides. Again, the general rule remains that the A′ cation should be larger than the B cation in this structural family.
In Fig. 16 we can see that although a broad range of existence appears for the m = 0, n = 1 members of this family, all other (m/n) members occupy a much more restricted phase space. The explanation for this may lie within the layers comprising the structures. The m = 0, n = 1 phases are built up exclusively from A3A′O6 layers, which seem to accommodate a large size range with respect to A′, which apparently leads to a lesser size restriction on the B cation. The other end member of this family, the 2H-perovskite, is comprised exclusively of A3O9 layers. In this case, the size of the B cation is greatly restricted and, consequently, only a handful of compositions are known to take on this structure type. It appears, that the inclusion of the A3O9 layers, which is the case for all compositions other than the m = 0, n = 1 members, leads to a restriction for the size of the B cation that the structure can accommodate and, hence, a more restricted phase space for all compositions having A3O9 layers. This is evidenced by the location of the A3A′BO6 compositions on the chart relative to the location of the 2H-perovskite oxides.
When examining the number of known compositions for each (m/n) phase, a precipitous drop is seen as the A3O9 layer is introduced (Table 2). The m = 0, n = 1 structure (no A3O9 layers) is the most common by far (115 compositions). The m = 1, n = 3 and m = 1, n = 1 phases are two other structures for which a significant number of compositions are known (24 and 14 compositions, respectively). One other interesting observation from the data shown in Table 2 is that Ca is limited almost exclusively to the m = 0, n = 1 structure. Ca, with its smaller size, seems to be far less stable in A3O9 layers than in the more size accommodating A3A′O6 layers, where it is the A cation for many compositions with m = 0, n = 1.
| Formula | m/n | A = Ca | A = Sr | A = Ba | All |
|---|---|---|---|---|---|
| A3A′BO6 | 0/1 | 31 | 65 | 7 | 115 |
| A15A′4B7O33 | 1/4 | 0 | 0 | 0 | 1 |
| A4A′B2O9 | 1/3 | 1 | 11 | 8 | 23 |
| A9A′2B5O21 | 1/2 | 0 | 4 | 1 | 5 |
| A14A′3B6O21 | 5/9 | 0 | 2 | 2 | 4 |
| A24A′5B14O57 | 3/5 | 0 | 1 | 0 | 1 |
| A5A′B3O12 | 2/3 | 0 | 1 | 2 | 3 |
| A16A′3B10O39 | 7/9 | 0 | 0 | 1 | 1 |
| A17A′3B11O42 | 8/9 | 0 | 0 | 1 | 1 |
| A6A′B4O15 | 1/1 | 0 | 5 | 8 | 14 |
| A7A′B5O18 | 4/3 | 0 | 0 | 3 | 3 |
| A8A′B6O21 | 5/3 | 0 | 0 | 2 | 2 |
| A9A′B7O24 | 2/1 | 0 | 0 | 2 | 2 |
| A32A′3B26O87 | 23/9 | 0 | 0 | 1 | 1 |
| A11A′B9O30 | 8/3 | 0 | 0 | 1 | 1 |
| A12A′B10O33 | 3/1 | 0 | 0 | 2 | 2 |
| A3B3O9 | 1/0 | 0 | 0 | 1 | 3 |
Sr and Ba are found in most other phases. While Sr most often forms the m = 0, n = 1 phase it readily forms A3O9 layers and hence is found in other (m/n) phases. Ba readily forms A3O9 layers and in fact, all the 2H-perovskite structures at ambient pressure contain Ba as the A cation. Consequently, it is not surprising that the (m/n) compositions that begin to approach the 2H-perovskite structure exclusively contain Ba.
When a tool such as this is compiled, there are often compositions that seem to defy the trend suggested by all others. If one observes the phase space for all members of this family where m ≠ 0, the composition Ba4NaMn2O946 is noteworthy. Not only is the mixed Mn valence intriguing, but also it is well outside the phase space suggested by related compositions. It was synthesized electrochemically, whereas most others have been made in fluxes or by the conventional solid-state method. This suggests that not only is there room for discovery within the borders observed, but there may well be room to push the boundaries further with the use of more exotic synthetic methods.59
There are three cations in the A3n + 3mA′nB3m + nO9m + 6n family of oxides and, hence, it stands to reason that there should be individual as well as overall relationships between them. Specifically, we expect a size correlation between the radius ratios of A and A′ due to the existence of A3A′O6 layers in the structure and we expect a size relationship between the B and A′ cations due to the existence of the one-dimensional chains of trigonal prisms and octahedra containing B and A′ cations, respectively. To incorporate these two relationships, we created a “reduced” plot of all three cations, by plotting the ratio of rA/rA′ against the ratio of rB/rA′. The result, shown in Fig. 17, indicates an overall radius ratio phase space for this family. As projected, the data suggest the trend that the closer in size the A′ cation is to A, the smaller the B cation must be. This is particularly obvious for the m = 0, n = 1 compositions. For all other m and n values, we again observe a much more restricted existence range. Not only are these compositions restricted to a narrow size distribution of the A′ and B cations, as shown in Fig. 16, but also there is an additional restriction placed upon them by the relative size of the A to the A′ cation sizes (and by extension by the B cation sizes). Hence, their existence phase space is circumscribed by a limiting range of the A, A′ and B cation radii, significantly restricting the selection of elemental compositions one can use to synthesize new compositions.
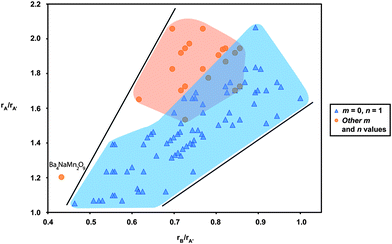 | ||
| Fig. 17 Reduced radius ratio plot of rA/rA′vs.rB/rA′ for the A3m + 3nA′nB3m + nO9m + 6n family of oxides. The region corresponding to the m = 1, n = 0 members is shaded in blue, while the region corresponding to all other m and n values is shaded in orange. The m = 0, n = 1 compositions can form for a wide range of reduced radii, while the compositions corresponding to other m and n values fall into a much more restricted region. | ||
Though simplistic at best, the plotting of ionic radii for the 2H-perovskite related oxide structure offers a simple tool for the synthetic chemist. Although plots created for other structure types have utilized other structurally relevant factors, the simplicity of this tool makes the utility for the synthetic chemist unparalleled. Using these diagrams, predictions for new compositions can readily be made and put to the test.
Acknowledgements
We gratefully acknowledge financial support from the National Science Foundation through grant DMR:0804209, from the Department of Energy, BES, through the Center for Heterogeneous Functional Materials for Energy Systems, award number DE-SC0001061, and from NASA via award number NNX10AN33A.References
- R. M. Mitchell, Perovskites: Modern and Ancient, Almaz Press, Thunder Bay, 2002 Search PubMed.
- A. L. Patterson and J. S. Kasper, International Tables for X-ray Crystallography, Vol. II, 1959, 342 Search PubMed.
- J. J. Randall and L. Katz, Acta Crystallogr., 1959, 12, 519 CrossRef CAS.
- K. E. Stitzer, J. Darriet and H.-C. zur Loye, Curr. Opin. Solid State Mater. Sci., 2001, 5, 535 CrossRef CAS.
- J. Darriet and M. A. Subramanian, J. Mater. Chem., 1995, 5, 543 RSC.
- J. M. Perez-Mato, G. Madariaga, F. J. Zuñiga and A. Garcia Arribas, Acta Crystallogr., Sect. A: Found. Crystallogr., 1987, 43, 216 CrossRef.
- J. M. Perez-Mato, M. Zakhour-Nakhl, F. Weill and J. Darriet, J. Mater. Chem., 1999, 9, 2795 RSC.
- R. S. Roth, J. Res. NBS, 1957, 58, 75 CAS.
- D. M. Giaquinta and H.-C. zur Loye, Chem. Mater., 1994, 6, 365 CrossRef CAS.
- D. Elwell and H. J. Scheel, Crystal Growth from High-Temperature Solutions, Academic Press, New York, 1975 Search PubMed.
- D. E. Bugaris and H.-C. zur Loye, Angew. Chem., Int. Ed., 2011 Search PubMed , in press.
- S. J. Mugavero III, W. R. Gemmill, I. P. Roof and H.-C. zur Loye, J. Solid State Chem., 2009, 182, 1950 CrossRef.
- P. D. Battle, G. R. Blake, J. C. Burley, E. J. Cussen, J. Sloan, J. F. Vente, J. Darriet and F. Weill, Solid State Chem. Inorg. Mater. II, 1999, 547, 45 CAS.
- M. Bharathy and H.-C. zur Loye, J. Chem. Crystallogr., 2010, 40, 945 CrossRef CAS.
- M. Bharathy, V. A. Rasolov and H.-C. zur Loye, Chem. Mater., 2008, 20, 2268 CrossRef CAS.
- P. D. Battle, S. J. Hartwell and C. A. Moore, Inorg. Chem., 2001, 40, 1716 CrossRef CAS.
- Y. Shi, Y. Guo, S. Yu, M. Arai, A. Sato, A. A. Belik, K. Yamaura and E. Takayama-Muromachi, J. Am. Chem. Soc., 2010, 132, 8474 CrossRef CAS.
- M. Bharathy, H. S. Khalsa, M. D. Smith and H.-C. zur Loye, Solid State Sci., 2009, 11, 294 CrossRef CAS.
- M. J. Davis, M. D. Smith, K. E. Stitzer and H.-C. zur Loye, J. Alloys Compd., 2003, 351, 95 CrossRef CAS.
- M. J. Davis, M. D. Smith and H.-C. zur Loye, Inorg. Chem., 2003, 42, 6980 CrossRef CAS.
- R. B. Macquart, W. R. Gemmill, M. J. Davis, M. D. Smith and H.-C. zur Loye, Inorg. Chem., 2006, 45, 4391 CrossRef CAS.
- T. Takami, H. Igarashi and M. Itoh, J. Phys. Soc. Jpn., 2010, 79, 044715 CrossRef.
- K. E. Stitzer, W. H. Henley, J. B. Claridge, H.-C. zur Loye and R. C. Layland, J. Solid State Chem., 2002, 164, 220 CrossRef CAS.
- M. J. Davis, M. D. Smith and H.-C. zur Loye, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2001, 57, 1234 CrossRef CAS.
- C. Lampe-Önnerud and H.-C. zur Loye, Inorg. Chem., 1996, 35, 2155 CrossRef.
- M. J. Davis, M. D. Smith and H.-C. zur Loye, J. Solid State Chem., 2003, 173, 122 CrossRef CAS.
- V. G. Zubkov, G. V. Bazuev, A. P. Tyutyunnik and I. F. Berger, J. Solid State Chem., 2001, 160, 293 CrossRef CAS.
- H. Fjellvåg, E. Gulbrandsen, S. Aasland, A. Olsen and B. C. Hauback, J. Solid State Chem., 1996, 124, 190 CrossRef.
- S. Aasland, H. Fjellvåg and B. Hauback, Solid State Commun., 1997, 101, 187 CrossRef CAS.
- A. Maignan, C. Michel, A. C. Masset, C. Martin and B. Raveau, Euro. Phys. J. B, 2000, 15, 657 CAS.
- W. Wong-Ng, G. Liu, J. Martin, E. L. Thomas, N. Lowhorn and J. A. Kaduk, J. Appl. Phys., 2010, 107, 033508 CrossRef.
- A. Jain, S. Singh and S. M. Yusuf, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 174419 CrossRef.
- I. Nowik, A. Jain, S. M. Yusuf and J. V. Yakhmi, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 054403 CrossRef.
- D. Flahaut, A. Maignan, S. Hébert, C. Martin, R. Retourx and V. Hardy, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 094418 CrossRef.
- X. X. Wang, J. J. Li, Y. G. Shi, Y. Tsujimoto, Y. F. Guo, S. B. Zhang, Y. Matsushita, M. Tanaka, Y. Katsuya, K. Kobayashi, K. Yamaura and E. Takayama-Muromachi, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 100410 CrossRef.
- K. Li, D. Sheptyakov, Y. Wang, C.-K. Loong and J. Lin, J. Solid State Chem., 2011, 184, 888 CrossRef CAS.
- W. T. A. Harrison, S. L. Hegwood and A. J. Jacobson, J. Chem. Soc., Chem. Commun., 1995, 19, 1953 RSC.
- J. Sun, G. Li, Z. Li, L. You and J. Lin, Inorg. Chem., 2006, 45, 8394 CrossRef CAS.
- K. E. Stitzer, A. El Abed, J. Darriet and H.-C. zur Loye, J. Am. Chem. Soc., 2001, 123, 8790 CrossRef CAS.
- K. Boulahya, M. Parras, J. M. González-Calbet and J. L. Martínez, Chem. Mater., 2004, 16, 5408 CrossRef CAS.
- M. Hernando, K. Boulahya, M. Parras, J. M. González-Calbet and U. Amador, Eur. J. Inorg. Chem., 2003, 2419 CrossRef CAS.
- A. El Abed, E. Gaudin and J. Darriet, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2002, 58, i138 CrossRef.
- C. A. Moore and P. D. Battle, J. Solid State Chem., 2003, 176, 88 CrossRef CAS.
- G. V. Bazuev, A. P. Tyutyunnik, I. F. Berger, I. V. Nikolaenko and B. G. Golovkin, J. Alloys Compd., 2011, 509, 6158 CrossRef CAS.
- A. El Abed, E. Gaudin, S. Lemaux and J. Darriet, Solid State Sci., 2001, 3, 887 CrossRef CAS.
- E. Quarez, P. Roussel, O. Pérez, H. Leligny, A. Bendraoua and O. Mentré, Solid State Sci., 2004, 6, 931 CrossRef CAS.
- K. Boulahya, M. Parras, J. M. González-Calbet and J. L. Martínez, Chem. Mater., 2003, 15, 3537 CrossRef CAS.
- K. Boulahya, M. Hernando, M. Parras and J. M. González-Calbet, J. Mater. Chem., 2007, 17, 1620 RSC.
- M. Hernando, K. Boulahya, M. Parras, A. Varela, J. L. Martínez and J. M. González-Calbet, Chem. Mater., 2002, 14, 4948 CrossRef CAS.
- J. N. Mwamuka, W. R. Gemmill, K. E. Stitzer, M. D. Smith and H.-C. zur Loye, J. Alloys Compd., 2004, 377, 91 CrossRef CAS.
- A. El Abed, E. Gaudin, H.-C. zur Loye and J. Darriet, Solid State Sci., 2003, 5, 59 CrossRef CAS.
- K. E. Stitzer, A. El Abed, J. Darriet and H.-C. zur Loye, J. Solid State Chem., 2004, 177, 1405 CrossRef CAS.
- K. E. Stitzer, A. El Abed, J. Darriet and H.-C. zur Loye, J. Am. Chem. Soc., 2004, 126, 856 CrossRef CAS.
- E. Mooser and W. B. Pearson, Acta Crystallogr., 1959, 12, 1015 CrossRef CAS.
- O. Muller and R. Roy, The Major Ternary Structural Families, Springer-Verlag, New York, 1974 Search PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751 CrossRef.
- V. A. Carlson and A. M. Stacy, J. Solid State Chem., 1992, 96, 332 CrossRef CAS.
- H. Kageyama, K. Yoshimura, K. Kosuge, M. Azuma, M. Takano, H. Mitamura and T. Goto, J. Phys. Soc. Jpn., 1997, 66, 3996 CrossRef CAS.
- T. N. Nguyen and H.-C. zur Loye, J. Cryst. Growth, 1997, 172, 183 CrossRef CAS.
- A. P. Wilkinson and A. K. Cheetham, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1989, 45, 1672 CrossRef.
- S. Frenzen and H. Müller-Buschbaum, Z. Naturforsch. B, 1996, 51b, 1204 Search PubMed.
- G. Wehrum and R. Hoppe, Z. Anorg. Allg. Chem., 1992, 617, 45 CrossRef CAS.
- R. F. Sarkozy, C. W. Moeller and B. L. Chamberland, J. Solid State Chem., 1974, 9, 242 CrossRef CAS.
- Y. Wang, D. Walker, B. H. Chen and B. A. Scott, J. Alloys Compd., 1999, 285, 98 CrossRef CAS.
- J. B. Claridge, R. C. Layland and H.-C. zur Loye, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1997, 53, 1740 Search PubMed.
- A. Tomaszewska and H. Müller-Buschbaum, Z. Anorg. Allg. Chem., 1992, 617, 23 CrossRef CAS.
- C. A. Moore, E. J. Cussen and P. D. Battle, J. Solid State Chem., 2000, 153, 254 CrossRef CAS.
- M. J. Davis, M. D. Smith, K. E. Stitzer and H.-C. zur Loye, J. Alloys Compd., 2003, 351, 95 CrossRef CAS.
- J. B. Claridge, R. C. Layland, W. H. Henley and H.-C. zur Loye, Z. Anorg. Allg. Chem., 1998, 624, 1951 CrossRef CAS.
- H. Kageyama, K. Yoshimura and K. Kosuge, J. Solid State Chem., 1998, 140, 14 CrossRef CAS.
- S. Niitaka, H. Kageyama, M. Kato, K. Yoshimura and K. Kosuge, J. Solid State Chem., 1999, 146, 137 CrossRef CAS.
- A. Tomaszewska and H. Müller-Buschbaum, Z. Anorg. Allg. Chem., 1993, 619, 534 CrossRef CAS.
- J. Darriet, F. Grasset and P. D. Battle, Mater. Res. Bull., 1997, 32, 139 CrossRef CAS.
- J. B. Claridge, R. C. Layland, R. D. Adams and H.-C. zur Loye, Z. Anorg. Allg. Chem., 1997, 623, 1131 CrossRef CAS.
- S. Kawasaki, M. Takano and T. Inami, J. Solid State Chem., 1999, 145, 302 CrossRef CAS.
- G. V. Bazuev, V. G. Zubkov, I. F. Berger and T. I. Arbuzova, Solid State Sci., 1999, 1, 365 CrossRef CAS.
- J. F. Vente, J. K. Lear and P. D. Battle, J. Mater. Chem., 1995, 5, 1785 RSC.
- M. D. Smith and H.-C. zur Loye, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2001, 57, 337 CrossRef CAS.
- T. N. Nguyen and H.-C. zur Loye, Neutron Scattering Mater. Sci., 1995, 376, 603 CAS.
- T. N. Nguyen and H.-C. zur Loye, J. Solid State Chem., 1995, 117, 300 CrossRef CAS.
- T. N. Nguyen, P. A. Lee and H.-C. zur Loye, Science, 1996, 271, 489 CAS.
- M. D. Smith and H.-C. zur Loye, Chem. Mater., 2000, 12, 2404 CrossRef CAS.
- R. C. Layland, S. L. Kirkland and H.-C. zur Loye, J. Solid State Chem., 1998, 139, 79 CrossRef CAS.
- P. Núñez, M. A. Rzeznik and H.-C. zur Loye, Z. Anorg. Allg. Chem., 1997, 623, 1269 CrossRef.
- M. James and J. P. Attfield, Chem.–Eur. J., 1996, 2, 737 CrossRef CAS.
- R. C. Layland, S. L. Kirkland, P. Núñez and H.-C. zur Loye, J. Solid State Chem., 1998, 139, 416 CrossRef CAS.
- N. Segal, J. F. Vente, T. S. Bush and P. D. Battle, J. Mater. Chem., 1996, 6, 395 RSC.
- B. A. Reisner and A. M. Stacy, J. Am. Chem. Soc., 1998, 120, 9682 CrossRef CAS.
- P. Núñez, S. Trail and H.-C. zur Loye, J. Solid State Chem., 1997, 130, 35 CrossRef.
- S. Frenzen and H. Müller-Buschbaum, Z. Naturforsch. B, 1995, 50, 581 CAS.
- T. N. Nguyen, D. M. Giaquinta and H.-C. zur Loye, Chem. Mater., 1994, 6, 1642 CrossRef CAS.
- W. H. Henley, J. B. Claridge, P. L. Smallwood and H.-C. zur Loye, J. Cryst. Growth, 1999, 204, 122 CrossRef CAS.
- M. D. Smith, J. K. Stalick and H.-C. zur Loye, Chem. Mater., 1999, 11, 2984 CrossRef CAS.
- A. V. Powell, P. D. Battle and J. G. Gore, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1993, 49, 852 CrossRef.
- G. Tams and H. Müller-Buschbaum, Z. Naturforsch. B, 1994, 49b, 581 Search PubMed.
- M. James and J. P. Attfield, J. Mater. Chem., 1994, 4, 575 RSC.
- R. C. Layland and H.-C. zur Loye, J. Alloys Compd., 2000, 299, 118 CrossRef CAS.
- C. Lampe-Önnerud, M. Sigrist and H.-C. zur Loye, J. Solid State Chem., 1996, 127, 25 CrossRef.
- M. Shimada, Y. Takeda, H. Taguchi, F. Kanamaru and M. Koizurmi, J. Cryst. Growth, 1975, 29, 75 CrossRef CAS.
- H. Taguchi, Y. Takeda, F. Kanamaru, M. Shimada and M. Koizumi, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1977, 33, 1299 CrossRef.
- E. J. Cussen and P. D. Battle, Chem. Mater., 2000, 12, 831 CrossRef CAS.
- J. J. Lander, Acta Crystallogr., 1951, 4, 148 CrossRef CAS.
- E. J. Cussen, J. F. Vente and P. D. Battle, J. Am. Chem. Soc., 1999, 121, 3958 CrossRef CAS.
- P. D. Battle, G. R. Blake, J. Darriet, J. G. Gore and F. Weill, J. Mater. Chem., 1997, 7, 1559 RSC.
- J. A. Campá, E. Gutiérrez-Puebla, M. A. Monge, I. Rasines and C. Ruíz-Valero, J. Solid State Chem., 1994, 108, 230 CrossRef.
- K. Boulahya, M. Parras and J. M. González-Calbet, J. Solid State Chem., 1999, 145, 116 CrossRef CAS.
- J. B. Claridge and H.-C. zur Loye, Chem. Mater., 1998, 10, 2320 CrossRef CAS.
- G. R. Blake, J. Sloan, J. F. Vente and P. D. Battle, Chem. Mater., 1998, 10, 3536 CrossRef CAS.
- J. A. Campá, E. Gutiérrez-Puebla, A. Monge, I. Rasines and C. Ruíz-Valero, J. Solid State Chem., 1996, 126, 27 CrossRef.
- J. Vacínová and J. L. Hodeau, J. Solid State Chem., 1998, 140, 201 CrossRef.
- F. Grasset, F. Weill and J. Darriet, J. Solid State Chem., 1998, 140, 194 CrossRef CAS.
- P. D. Battle, G. R. Blake, J. Sloan and J. F. Vente, J. Solid State Chem., 1998, 136, 103 CrossRef CAS.
- J. Lee and G. F. Holland, J. Solid State Chem., 1991, 93, 267 CrossRef CAS.
- F. Abraham, S. Minaud and C. Renard, J. Mater. Chem., 1994, 4, 1763 RSC.
- K. E. Stitzer, M. D. Smith, J. Darriet and H.-C. zur Loye, Chem. Commun., 2001, 1680 RSC.
- K. Boulahya, M. Parras and J. M. González-Calbet, J. Solid State Chem., 1999, 142, 419 CrossRef CAS.
- H.-C. zur Loye, K. E. Stitzer, M. D. Smith, A. El Abed and J. Darriet, Inorg. Chem., 2001, 40, 5152 CrossRef CAS.
- O. Gourdon, V. Petricek, M. Dusek, P. Bezdicka, S. Durovic, D. Gyepesova and M. Evain, Acta Crystallogr., Sect. B: Struct. Sci., 1999, 55, 841 Search PubMed.
- A. Schönleber, F. J. Zuniga, J. M. Perez-Mato, J. Darriet and H.-C. zur Loye, Acta Crystallogr., Sect. B: Struct. Sci., 2006, 62, 197 Search PubMed.
- M. Zakhour-Nakhl, J. B. Claridge, J. Darriet, F. Weill, H.-C. zur Loye and J. M. Perez-Mato, J. Am. Chem. Soc., 2000, 122, 1618 CrossRef CAS.
- P. D. Battle, J. C. Burley, E. J. Cussen, J. Darriet and F. Weill, J. Mater. Chem., 1999, 9, 479 RSC.
- G. V. Bazuev, N. A. Zaitseva and D. G. Kellerman, Solid State Sci., 2003, 5, 1465 CrossRef CAS.
- M. Zakhour-Nakhl, F. Weill, J. Darriet and J. M. Perez-Mato, Int. J. Inorg. Mater., 2000, 2, 71 CrossRef CAS.
- N. A. Jordan, P. D. Battle, S. van Smaalen and M. Wunschel, Chem. Mater., 2003, 15, 4262 CrossRef CAS.
- M. Zakhour-Nakhl, J. Darriet, J. B. Claridge, H.-C. zur Loye and J. M. Perez-Mato, Int. J. Inorg. Mater., 2000, 2, 503 CrossRef CAS.
- M. Evain, F. Boucher, O. Gourdon, V. Petricek, M. Dusek and P. Bezdicka, Chem. Mater., 1998, 10, 3068 CrossRef CAS.
- T. K. Mandal, A. M. Abakumov, J. Hadermann, G. V. Tendeloo, M. Croft and M. Greenblatt, Chem. Mater., 2007, 19, 6158 CrossRef CAS.
- T. Shishido, Y. Saito, N. Toyota, K. Ukei, T. Sasaki and T. Fukuda, Mol. Cryst. Liq. Cryst., 1990, 184, 177 CrossRef CAS.
- Y. Saito, T. Shishido, N. Toyota, K. Ukei, T. Sasaki and T. Fukuda, J. Cryst. Growth, 1991, 109, 426 CrossRef CAS.
- K. Ukei and A. Yamamoto, Acta Crystallogr., Sect. B: Struct. Sci., 1993, 49, 67 CrossRef.
- A. M. Abakumov, A. V. Mironov, V. A. Govorov, M. V. Lobanov, M. G. Rozova, E. V. Antipov, O. I. Lebedev and G. V. Tendeloo, Solid State Sci., 2003, 5, 1117 CrossRef CAS.
- G. V. Bazuev, V. N. Krasil'nikov and D. G. Kellerman, J. Alloys Compd., 2003, 352, 190 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |