 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemoenzymatic preparation of germacrene analogues†
Oscar
Cascón
,
Sabrina
Touchet
,
David J.
Miller
,
Veronica
Gonzalez
,
Juan A.
Faraldos
and
Rudolf K.
Allemann
*
School of Chemistry and Cardiff Catalysis Institute, Cardiff University, Main Building, Park Place, Cardiff CF10 3AT, UK. E-mail: allemannrk@cardiff.ac.uk; Fax: +44 (0)29 208 74030; Tel: +44 (0)29 208 79014
First published on 15th August 2012
Abstract
A small library of novel germacrenes was generated using a combination of two plant enzymes, germacrene A synthase, and D synthase and modified farnesyl diphosphate (FDP) analogues. This chemoenzymatic approach allows the preparation of potentially valuable volatiles for biological studies.
Terpenoids represent a valuable class of bioactive fine chemicals1 and are therefore attractive targets for synthetic modification; modulation of their natural properties may lead to new medicinal and agrochemical compounds with improved properties. However, the complexity of the hydrocarbon skeletons and the often significant chemical instability of many terpenoids can present a formidable challenge to the synthetic chemist.2 Synthetic biology approaches have focused on the preparation of natural terpenoids in living organisms,3a but they operate with whole biochemical pathways using fundamental biosynthetic building blocks (e.g. isopentenyl diphosphate) and can therefore not easily be applied to generate modified terpenes. One attractive synthetic approach that complements current terpene synthetic biology3b,c and circumvents the difficult task of engineering full metabolic pathways to generate alternative substrates in vivo, could rely on the chemical preparation of FDP analogues as substrates of recombinant terpene synthases to create modified terpenoids. Modified FDPs4,5 have been used extensively to study the mechanisms of the cationic reactions mediated by (sesqui)terpene synthases.4–7 However, despite the fact that several unnatural FDPs are indeed turned over by these enzymes,4,5,8 only a few reports have explored the synthetic utility of terpene synthases toward the production of valuable novel terpenoids.9
Germacrene A and germacrene D synthases (GAS and GDS) are two plant sesquiterpene synthases that catalyze the Mg2+-dependent conversion of FDP (1a) to germacrene A (3a) and germacrene D (5a), respectively (Fig. 1). These two macrocyclic sesquiterpenes have been shown to act as semiochemicals affecting the olfactory response of insects.10 While a synthesis of the rather unstable germacrene D (5a) has been reported,11 the extreme thermal and photochemical instability of the acid labile germacrene A (3a) has so far hampered the development of a satisfactory chemical synthesis.12 Nevertheless, fluorinated germacrene A analogues with improved stabilities have previously been produced enzymatically from fluorinated FDP analogues.4a,c,5b
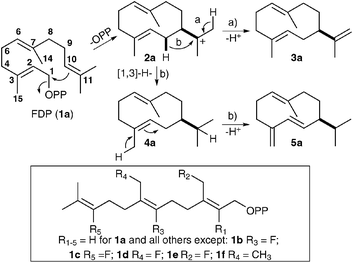 | ||
| Fig. 1 Proposed biosynthesis of germacrenes A (3a) and D (5a). Modified substrate analogues of GAS and GDS (framed). | ||
Thus, based on the biological and potential economic significance of compounds 3a and 5a, germacrene A and D synthases from Solidago canadensis13 were selected to investigate their capability to produce non-natural germacrenes from modified FDPs. To this end, recombinant GAS and GDS were overproduced in E. coli and purified as previously described.9b,14 Several fluorine and methyl modified FDPs were screened by GC-MS on an analytical scale for substrate activity. Germacrene A analogues were readily identified through their ability to undergo thermal Cope rearrangements to the corresponding β-elemene analogues under GC-MS conditions;14 germacrene D analogues were identified from their mass spectra since the presence of the more stable (i.e. more abundant, 100%) [M − 43]+ fragment in the EI+-MS is diagnostic of the parent 5a. Only modified FDP analogues (Fig. 1, framed) that gave a relatively strong ion count in the total ion chromatogram (GC-MS) as compared with the natural substrate 1a were considered suitable for this study (vide infra).
Interestingly, in contrast to what has been observed with other sesquiterpene synthases,4a,d,9a 2-fluoro-FDP was not turned over significantly by GAS or GDS. In addition, the H/F and in particular the H/CH3 substitution at the C15 position of FDP was not tolerated by either enzyme. The full details and the mechanistic implications of these observations are beyond the scope of this manuscript and will be published elsewhere.
Optimal reaction conditions for the preparation of modified germacrenes: initial preparative incubations using both enzymes were shown to be inefficient and hence an optimisation of the reaction conditions was carried out. After some experimentation, conversions were found to be optimal at concentrations of Mg2+, FDP and enzyme of 10 mM (5 mM for GAS), 0.35 mM and 6 μM, respectively (ESI†). Higher concentrations of Mg2+ and/or enzyme led to the formation of insoluble/inactive FDP–Mg2+-complexes and/or enzyme aggregation, which in turn resulted in less efficient turnovers. The concentration of GDS (but not GAS) could be increased to 12 μM simply by inclusion of 1% of the non-denaturing detergent 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate) (CHAPS) in the assay buffer. The nature of the organic solvent, the reaction vessel itself and the extractive work up were of fundamental importance for optimal conversions.9b In the final optimised conditions, d-chloroform was used as the organic layer, the incubations were carried out in sealed tubes with gentle agitation and enzymatic products were extracted overnight using an automated rotator. The filtered and dried d-chloroform solutions were then analysed by GC-MS and NMR-spectroscopy. Under these conditions, the enzymatic conversions of 1a to germacrene D (5a) and germacrene A (3a) were 76% and 40%, respectively (Fig. 2).
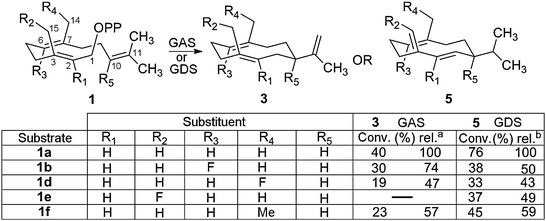 | ||
| Fig. 2 Incubations of modified FDP (1b, d–f) under optimized conditions: a GAS (6 μM), Mg2+ (5 mM) and FDP (0.35 mM); b GDS (12 μM), Mg2+ (10 mM) and FDP (0.35 mM). Conversions were determined by GC-FID (ESI†) in pentane. Relative conversions (rel.) denote percentage with respect to 1a. | ||
Incubations of FDP analogues with GAS: fluorinated germacrene A analogues were obtained from incubations of GAS with 6F-FDP (1b) and 14F-FDP (1d) (Fig. 2). These compounds were identified by GC-MS through co-elution with authentic material previously isolated from experiments with aristolochene synthase from Penicillium roqueforti (PR-AS, see ESI†).4c
Upon incubation with GAS, 10F-FDP (1c) produced efficiently (30% rel.) a single fluorinated hydrocarbon that co-eluted in the GC-MS column with an authentic sample of α-10F-humelene, a known compound prepared previously using δ-cadinene synthase (DCS).4d This result demonstrates that with diphosphate 1c, both enzymes are able to catalyze an anti-Markovnikov 1,11-macrocyclisation via π-donation from the vinylic fluorine atom into the distal C10,C11-double bond of 1c.
14Me-FDP (1f) was also readily turned over by GAS yielding a mixture of at least seven hydrocarbons. The major product (retention time 28.5 min, approx. >50% total hydrocarbons, ESI†) underwent a thermal rearrangement, thereby suggesting a germacrene A analogue as the major enzymatic product from substrate 1f. In addition, in a parallel study with PR-AS, the same compound mixture was generated from 1f (ESI†). Purification by preparative TLC and subsequent 1H-NMR analysis of this sample unambiguously confirmed the structure of the major GAS- and PR-AS-generated products as 14Me-germacrene A (3f). Hydrocarbon 3f displayed the well documented conformational flexibility exhibited by germacrene A (3a).12 Indeed, 1H-NMR spectra comparisons (ESI†) with those previously obtained for (3a) at different temperatures12 suggested that 3d exists as an interconverting mixture of the same three conformers observed for 3a, albeit with different relative populations (ESI†). As with the parent hydrocarbon 3a, the most abundant conformation corresponds to the ‘crossed up–up’ (UU) configuration,12 but in contrast to 3a, the combined ‘parallel down–up’ DU and ‘up–down’ UD conformations of 3f dominates (61%) over the individually more stable (i.e. more abundant) UU conformation (39%). The present conformational distribution relates to the apparent increase in steric bulk on C14 of 3f relative to 3a, which likely raises the energy of the UU conformer with respect to the UD and DU conformations. For a diagram and further explanation regarding the conformations of 3f see ESI.†
Incubations of FDP analogues with GDS: 6F-FDP (1b) and 14Me-FDP (1f) were turned over efficiently by GDS each giving a single product displaying the more abundant and stable [M − 43]+ fragment in their EI+-MS spectra, which is characteristic of germacrene D (5a) through loss of the isopropyl group within the MS-detector. Indeed, preparative incubations followed by direct 1H- and 19F-NMR spectroscopy analysis confirmed their identity as the expected germacrene D analogues (ESI†).
Two products (10% rel.) in an approximate 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio were observed in the pentane extracts from incubation of 10F-FDP (1c) with GDS (ESI†). GC-MS analysis of a mixture of 10-fluoro-farnesenes, prepared in a previous study,4d unambiguously identified the major product as (E)-β-10F-farnesene (ESI†). Interestingly, the minor component (25%) of this mixture was also identified by co-elution as the fluorinated α-10F-humulene previously observed in incubations of 1c with GAS or DCS.4d
:1 ratio were observed in the pentane extracts from incubation of 10F-FDP (1c) with GDS (ESI†). GC-MS analysis of a mixture of 10-fluoro-farnesenes, prepared in a previous study,4d unambiguously identified the major product as (E)-β-10F-farnesene (ESI†). Interestingly, the minor component (25%) of this mixture was also identified by co-elution as the fluorinated α-10F-humulene previously observed in incubations of 1c with GAS or DCS.4d
Both 15F-FDP (1e) and 14F-FDP (1d) were converted by GDS as judged by GC-MS to a well-defined product under analytical conditions; each product (5e and 5d) displayed the major [M − 43]+-fragment suggesting that they were indeed germacrene D derivatives. However, prolonged preparative incubations led to the formation of a second product apparently arising from the initial GDS-generated product (ESI†). Although the presence of this minor product hampered a full NMR interpretation of the spectrum of the original enzymatic product (5e), the observation (1H NMR, 500 MHz) of a relatively downfield (approx. 2 ppm with respect to 5a) wide doublet at δH = 6.53 ppm (2JH–F = 86.0 Hz, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHF) instead of the diagnostic broad doublet at δH = 4.77 ppm (2JH–H = 13.0 Hz, CCHH, exo methylene group) of 5a13c (ESI†) is consistent with the major product being 15F-germacrene D (5e, Fig. 2). Surprisingly, the 19F-NMR spectrum of this mixture displayed three absorbances, two identical doublets (2JH–F = 86.0 Hz) at δF = −136.1 (minor) and −138.6 (major) ppm, respectively, plus a downfield triplet (2JH–F = 46.0 Hz) at −184.8 ppm due to the very minor peak observable by GC-MS. Thus, the major (and only) enzymatic product is most likely produced by GDS as a mixture of two geometric 15F-germacrene D isomers (5e) (Fig. 2) that is not resolved by GC-MS. This observation implies that the corresponding tightly bound carbocation (4e) possesses sufficient mobility within the active site of GDS to allow a not completely specific proton-loss to generate the observed isomeric mixture of 5e.
CHF) instead of the diagnostic broad doublet at δH = 4.77 ppm (2JH–H = 13.0 Hz, CCHH, exo methylene group) of 5a13c (ESI†) is consistent with the major product being 15F-germacrene D (5e, Fig. 2). Surprisingly, the 19F-NMR spectrum of this mixture displayed three absorbances, two identical doublets (2JH–F = 86.0 Hz) at δF = −136.1 (minor) and −138.6 (major) ppm, respectively, plus a downfield triplet (2JH–F = 46.0 Hz) at −184.8 ppm due to the very minor peak observable by GC-MS. Thus, the major (and only) enzymatic product is most likely produced by GDS as a mixture of two geometric 15F-germacrene D isomers (5e) (Fig. 2) that is not resolved by GC-MS. This observation implies that the corresponding tightly bound carbocation (4e) possesses sufficient mobility within the active site of GDS to allow a not completely specific proton-loss to generate the observed isomeric mixture of 5e.
In summary, the results presented here provide insight into aspects of the reaction mechanisms employed by GAS and GDS and describe a general chemoenzymatic approach for the synthesis of non-natural terpenoids that are otherwise not easily accessible by classical chemical synthesis or synthetic biology. Indeed, these results show that GAS and GDS can turn over a variety of modified FDPs to germacrene A and D analogues often with synthetically acceptable conversions and in sufficient amounts for biological testing as semiochemicals.
This work was supported by the United Kingdom's Biotechnology and Biological Sciences Research Council through grants BB/G003572/1 and BB/H01683X/1 and by Cardiff University. We thank Dr Rob Jenkins, Cardiff University, for assistance with NMR spectroscopy.
Notes and references
- J. S. Glasby, Encyclopedia of Terpenoids, Wiley, Chichester, 1982 Search PubMed.
- A. J. Minnaard, J. B. P. A. Wijnberg and A. De Groot, Tetrahedron, 1999, 55, 2115–2146 CrossRef CAS.
- (a) N. Misawa, Curr. Opin. Biotechnol., 2011, 22, 627–633 CrossRef CAS; (b) P. A. Ajikumar, et. al. , Science, 2010, 330, 70–74 CrossRef CAS; (c) P. J. Westfall, et. al. , Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 111–118 CrossRef.
- (a) D. J. Miller, F. L. Yu and R. K. Allemann, ChemBioChem, 2007, 8, 1819–1825 CrossRef CAS; (b) D. J. Miller, F. L. Yu, N. J. Young and R. K. Allemann, Org. Biomol. Chem., 2007, 5, 3287–3298 RSC; (c) D. J. Miller, F. L. Yu, D. W. Knight and R. K. Allemann, Org. Biomol. Chem., 2009, 7, 962–975 RSC; (d) J. A. Faraldos, D. J. Miller, V. Gonzalez, Z. Yoosuf-Aly, O. Cascón, A. Li and R. K. Allemann, J. Am. Chem. Soc., 2012, 134, 5900–5908 CrossRef CAS.
- (a) J. P. Noel, N. Dellas, J. A. Faraldos, M. Zhao, B. A. Hess, L. Smentek, R. M. Coates and P. E. O'Maille, ACS Chem. Biol., 2010, 5, 377–392 CrossRef CAS; (b) J. A. Faraldos, Y. X. Zhao, P. E. O'Maille, J. P. Noel and R. M. Coates, ChemBioChem, 2007, 8, 1826–1833 CrossRef CAS; (c) Y. Jin, D. C. Williams, R. Croteau and R. M. Coates, J. Am. Chem. Soc., 2005, 127, 7834–7842 CrossRef CAS; (d) D. E. Cane, G. H. Yang, Q. Xue and J. H. Shim, Biochemistry, 1995, 34, 2471–2479 CrossRef CAS; (e) D. E. Cane and Y. S. Tsantrizos, J. Am. Chem. Soc., 1996, 118, 10037–10040 CrossRef CAS.
- (a) D. J. Miller and R. K. Allemann, Nat. Prod. Rep., 2012, 29, 60–71 RSC; (b) E. Y. Shishova, F. L. Yu, D. J. Miller, J. A. Faraldos, Y. X. Zhao, R. M. Coates, R. K. Allemann, D. E. Cane and D. W. Christianson, J. Biol. Chem., 2008, 283, 15431–15439 CrossRef CAS; (c) F. L. Yu, D. J. Miller and R. K. Allemann, Chem. Commun., 2007, 40, 4155–4157 RSC; (d) H. A. Gennadios, V. Gonzalez, L. Di Costanzo, A. Li, F. L. Yu, D. J. Miller, R. K. Allemann and D. W. Christianson, Biochemistry, 2009, 48, 6175–6183 CrossRef CAS; (e) J. A. Aaron, X. Lin, D. E. Cane and D. W. Christianson, Biochemistry, 2010, 49, 1787–1797 CrossRef CAS; (f) R. P. McAndrew, P. P. Peralta-Yahya, A. De Giovanni, J. H. Pereira, M. Z. Hadi, J. D. Keesling and P. D. Adams, Structure, 2011, 19, 1876–1884 CrossRef CAS; (g) J. A. Faraldos, A. K. Antonczak, V. González, R. Fullerton, E. M. Tippmann and R. K. Allemann, J. Am. Chem. Soc., 2011, 133, 13906–13909 CrossRef CAS.
- (a) D. W. Christianson, Chem. Rev., 2006, 106, 3412–3442 CrossRef CAS; (b) M. Koksal, Y. H. Jin, R. M. Coates, R. Croteau and D. W. Christianson, Nature, 2011, 469, 116–120 CrossRef; (c) M. Koksal, H. Y. Hu, R. M. Coates, R. J. Peters and D. W. Christianson, Nat. Chem. Biol., 2011, 7, 431–433 CrossRef.
- (a) J. A. Faraldos, B. Kariuki and R. K. Allemann, J. Org. Chem., 2010, 75, 1119–1125 CrossRef CAS; (b) J. A. Faraldos and R. K. Allemann, Org. Lett., 2011, 13, 1202–1205 CrossRef CAS; (c) J. A. Faraldos, B. M. Kariuki and R. M. Coates, Org. Lett., 2011, 13, 836–839 CrossRef CAS.
- (a) L. S. Vedula, Y. X. Zhao, R. M. Coates, T. Koyama, D. E. Cane and D. W. Christianson, Arch. Biochem. Biophys., 2007, 466, 260–266 CrossRef CAS; (b) Z. Yoosuf-Aly, J. A. Faraldos, D. J. Miller and R. K. Allemann, Chem. Commun., 2012, 48, 7040–7042 RSC.
- (a) W. S. Bowers, C. Nishino, M. W. Nielson, M. E. Montgomery and L. R. Nault, Science, 1977, 196, 680–681 CAS; (b) R. Mozuraitis, M. Stranden, M. I. Ramirez, A. K. Borg-Karlson and H. Mustaparta, Chem. Sens., 2002, 27, 505–509 CrossRef CAS; (c) T. J. A. Bruce, et. al. , Pest Manage. Sci., 2005, 61, 1115–1121 CrossRef CAS; (d) M. A. Birkett, et. al. , Phytochemistry, 2008, 69, 1710–1715 CrossRef CAS.
- S. L. Schreiber and R. C. Hawley, Tetrahedron Lett., 1985, 26, 5971–5974 CrossRef CAS.
- J. A. Faraldos, S. Wu, J. Chappell and R. M. Coates, Tetrahedron, 2007, 63, 7733–7742 CrossRef CAS.
- (a) I. Prosser, A. L. Phillips, S. Gittings, M. J. Lewis, A. M. Hooper, J. A. Pickett and M. H. Beale, Phytochemistry, 2002, 60, 691–702 CrossRef CAS; (b) C. O. Schmidt, H. J. Bouwmeester, S. Franke and W. A. Konig, Chirality, 1999, 11, 353–362 CrossRef CAS; (c) N. Bulow and W. A. Koning, Phytochemistry, 2000, 55, 141–168 CrossRef CAS.
- M. J. Calvert, P. R. Ashton and R. K. Allemann, J. Am. Chem. Soc., 2002, 124, 11636–11641 CrossRef CAS.
- A. M. Adio, Tetrahedron, 2009, 65, 1533–1552 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic methods, enzyme production and purification, kinetic data, analytical data of reaction products. See DOI: 10.1039/c2cc35542f |
| This journal is © The Royal Society of Chemistry 2012 |