 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diastereoselective synthesis of half-sandwich chiral-at-metal cobaltacycles by oxidative cyclisation†
Jahangir
Amin
and
Christopher J.
Richards
*
School of Chemistry, University of East Anglia, Norwich Research Park, Norwich, NR4 7TJ, UK. E-mail: Chris.Richards@uea.ac.uk; Fax: +44 (0)1603 592003; Tel: +44 (0)1603 592003
First published on 10th August 2012
Abstract
Reaction of chiral ester linked diynes with chlorotris(triphenylphosphine)cobalt(I) and sodium cyclopentadienide gave (η5-cyclopentadienyl)(triphenylphosphine) cobaltacyclopentadiene complexes as single chiral-at-metal diastereoisomers, including a non-racemic example synthesised in three steps from (S)-3-butyn-2-ol.
The variability in the structure of chiral organometallic complexes provides novel opportunities for the synthesis of non-racemic ligands, catalysts and materials. Ideally such complexes are air-stable, configurationally-stable, and readily generated as single enantiomers. Chiral organometallics result from the attachment of a metal to a carbon based stereogenic centre (Fig. 1, A).1 Alternatively, differential di-substitution of metallocenes such as ferrocene (B) gives rise to planar chirality, these and other sandwich complexes having been exploited extensively in chiral ligand syntheses.2 Pseudo tetrahedral half-sandwich complexes containing three different additional ligands, with either an η5-cyclopentadienyl (C) or an η6-arene ligand, contain a stereogenic metal atom.3 Although such complexes have not been utilised as building blocks for ligand synthesis, this metal-focused chirality has been exploited extensively in stoichiometric asymmetric synthesis (notably M = Re, Fe, Mo),4 and more recently in catalysis.5
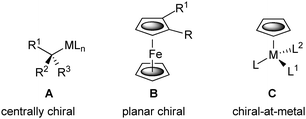 | ||
| Fig. 1 Representative chiral organometallic complexes. | ||
Methods for the generation of enantiomerically pure chiral-at-metal half-sandwich complexes began with resolution6 and have been extended to diastereoselective protocols mediated either by a chiral η5 or η6 π-ligand, or by an introduced mono or bidentate chiral ligand.3 Known examples of η5-cyclopentadienyl cobalt(III) complexes of type C have been generated by the latter procedure following ligand substitution with both neutral and anionic chiral ligands.7 In this Communication we report on an alternative oxidative cyclisation protocol for the highly diastereoselective and modular synthesis of cobalt-based chiral half-sandwich complexes, and on the extension of this methodology to the asymmetric synthesis of an air and configurationally stable chiral-at-cobalt complex.
Cobaltacyclopentadiene 1 is readily prepared from the reaction of chlorotris(triphenylphosphine)cobalt(I) and sodium cyclopentadienide with two equivalents of diphenylacetylene.8 The same reaction on a diyne containing a stereogenic centre within an acetylene tether will result in a chiral-at-metal cobaltacycle 2 (Scheme 1). Provided the reaction is diastereoselective, and the product configurationally stable, this will provide an accessible route to novel chiral organometallic building blocks.
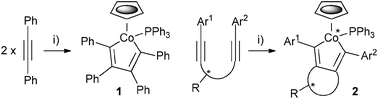 | ||
| Scheme 1 Known (1) and proposed (2) products of oxidative cyclisation. Reagents and conditions: (i) NaCp, CoCl(PPh3)3, Δ. | ||
Non-terminal linked diynes were prepared in two steps by an esterification and Sonogashira cross-coupling sequence; starting either from propargylic alcohols 3 and introduction of Ar2C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C– onto 4, or from 5 and introduction of Ar1 onto 6 (Scheme 2). Diynes 7 were chosen for this study because of the simplicity and modularity of these procedures, and also because they are known to react with (η5-cyclopentadienyl)cobaltdicarbonyl to give planar chiral (η5-cyclopentadienone)(η5-cyclopentadienyl)cobalt metallocenes in moderate diastereoselectivity.9
C– onto 4, or from 5 and introduction of Ar1 onto 6 (Scheme 2). Diynes 7 were chosen for this study because of the simplicity and modularity of these procedures, and also because they are known to react with (η5-cyclopentadienyl)cobaltdicarbonyl to give planar chiral (η5-cyclopentadienone)(η5-cyclopentadienyl)cobalt metallocenes in moderate diastereoselectivity.9
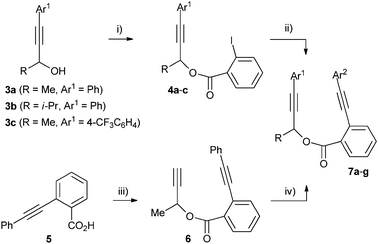 | ||
| Scheme 2 Diyne syntheses. Reagents and conditions: (i) 2-iodobenzoic acid (1.1 eq.), DCC (1.1 eq.), DMAP (0.2 eq.), CH2Cl2, RT, 48 h 92–99%. (ii) Ar2CCH (1.1 eq.), PdCl2(PPh3)2 (0.03 eq.), CuI (0.1 eq.), NEt3, 60 °C, 24 h, 93–99%. (iii) 3-butyn-2-ol (1 eq.), DCC (1.1 eq.), DMAP (0.2 eq.), CH2Cl2, RT, 48 h, 94%. (iv) Ar1I (1.1 eq.), PdCl2(PPh3)2 (0.03 eq.), CuI (0.1 eq.), NEt3, 60 °C, 24 h, 99%. | ||
Reaction of 7a (R = Me, Ar1 = Ar2 = Ph) with chlorotris(triphenylphosphine)cobalt(I) and sodium cyclopentadienide in THF heated at reflux for 30 minutes resulted in a new air-stable organometallic 8a isolated in 44% yield following column chromatography (Scheme 3, Table 1, Method A – entry 1). As the reaction likely proceeds via the in situ formation of (η5-cyclopentadienyl)cobaltbis(triphenylphosphine), pre-formation and isolation of this complex10 was followed by addition of 7a and heating in THF as before to give 8a in 75% yield (Method B – entry 2).
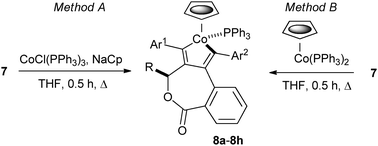 | ||
| Scheme 3 Diastereoselective synthesis of half-sandwich complexes 8. | ||
| Entry/diyne | R | Ar1 | Ar2 | Method | Product/yielda (%) |
|---|---|---|---|---|---|
| a Isolated by column chromatography. | |||||
| 1 7a | Me | Ph | Ph | A | 8a 44 |
| 2 7a | Me | Ph | Ph | B | 8a 75 |
| 3 7b | i-Pr | Ph | Ph | A | 8b 55 |
| 4 7b | i-Pr | Ph | Ph | B | 8b 79 |
| 5 7c | Me | 4-CF3C6H4 | Ph | A | 8c 78 |
| 6 7d | Me | Ph | 4-CF3C6H4 | A | 8d 72 |
| 7 7e | Me | Ph | 3-C5H4N | B | 8e 89 |
| 8 7f | Me | 2-BrC6H4 | Ph | A | 8f 73 |
| 9 7g | Me | 2-Cl-5-C5H3N | Ph | B | 8g 78 |
Examination of both crude and column isolated 8a by 1H NMR spectroscopy revealed four sets of signals in a 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5:1:1 ratio. Following recrystallisation of 8a the same ratio of signals was observed when the spectrum was recorded within minutes of dissolving the crystals in CDCl3 at room temperature (20 °C). No change in this ratio was observed over time. The multiplicity of signals in the 1H NMR pointed to the possibility that these may, in part, result from rapid epimerisation of the metal-based stereogenic centre.11 Stereochemical lability in η5-cyclopentadienyl piano-stool complexes C is a consequence of facile ligand dissociation.3 For example, epimerisation of the related isoelectronic chiral-at-metal complex (η5-cyclopentadienyl)FeCH3(CO)PPh2R* (half-life 70 min at 70 °C) proceeds by phosphine dissociation and formation of a planar 16-electron intermediate.12 Addition of 1.5 eq. of tri(p-tolyl)phosphine to 8a in CDCl3 at room temperature and recording the 1H NMR spectrum after 1 h revealed the presence of only 8a/tri(p-tolyl)phosphine and no new ligand substitution complex. An X-ray structure analysis of 8a confirmed the anticipated cobaltacycle half-sandwich structure, and revealed the relative configuration as SCo*, SC* (Fig. 2).13
:1.5:1:1 ratio. Following recrystallisation of 8a the same ratio of signals was observed when the spectrum was recorded within minutes of dissolving the crystals in CDCl3 at room temperature (20 °C). No change in this ratio was observed over time. The multiplicity of signals in the 1H NMR pointed to the possibility that these may, in part, result from rapid epimerisation of the metal-based stereogenic centre.11 Stereochemical lability in η5-cyclopentadienyl piano-stool complexes C is a consequence of facile ligand dissociation.3 For example, epimerisation of the related isoelectronic chiral-at-metal complex (η5-cyclopentadienyl)FeCH3(CO)PPh2R* (half-life 70 min at 70 °C) proceeds by phosphine dissociation and formation of a planar 16-electron intermediate.12 Addition of 1.5 eq. of tri(p-tolyl)phosphine to 8a in CDCl3 at room temperature and recording the 1H NMR spectrum after 1 h revealed the presence of only 8a/tri(p-tolyl)phosphine and no new ligand substitution complex. An X-ray structure analysis of 8a confirmed the anticipated cobaltacycle half-sandwich structure, and revealed the relative configuration as SCo*, SC* (Fig. 2).13
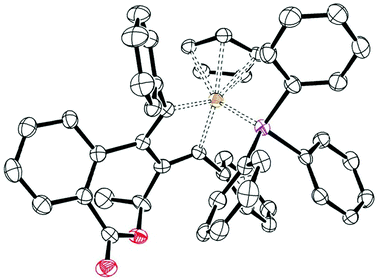 | ||
| Fig. 2 X-ray crystal structure of SCo*, SC* 8a. | ||
That facile phosphine substitution is not occurring with 8a reveals that the SCo*, SC* configuration is maintained in solution and that a single chiral-at-metal diastereoisomer results from oxidative cyclisation via an intermediate planar14 16 electron (η5-cyclopentadienyl)cobaltacyclopentadiene. Coordination of triphenylphosphine opposite the methyl group dictates the configuration of the metal-based chirality.
The solution isomerism is ascribed to the two other elements of chirality present in 8a. Three-atom linked biphenyls 9 interconvert rapidly between atropisomers15 containing either an equatorial or axial R substituent, the lowest energy arrangement being dependent upon the identity of X, Y and R (Scheme 4).16 The X-ray structure of 8a reveals an axial methyl group and an Ra* configuration with the cobaltacyclopentadiene moiety replacing the bottom phenyl group of 9. The propeller-like arrangement of the phenyl rings of a metal-coordinated triphenylphosphine complex 10 result in M and P-configurations which give rise to diastereoisomers with chiral-at-metal half-sandwich complexes.17 Although the barrier to intramolecular interconversion is usually low, occurring by a two ring-flip mechanism,17a,18 isolable M and P epimers have been obtained with a bulky chiral bidentate ligand, epimerisation occurring via reversible phosphine dissociation.19 Further examination of the X-ray structure reveals an M* configuration,20 and assuming the maintenance of this Ra*, M*-structure as the dominant species in solution, isomerisation gives rise to the three minor isomers listed in Scheme 4.21 In contrast, the isopropyl substituted complex 8b, prepared by both Methods A and B (entries 3 and 4), resulted in only a single observable stereoisomer in solution, a consequence of the greater conformational control imparted by the larger isopropyl group.
 | ||
| Scheme 4 Isomerism of 9, 10 and extension to SCo*, SC* 8a. | ||
A number of other complexes were prepared in good yield (8c–8g, entries 5–9), including examples with pyridyl ligand substituents (8e, 8g), and a complex with a 2-bromophenyl substituent (8f) with the potential for further functionalisation. Like parent methyl substituted complex 8a, all of these gave four solution species with one dominant (e.g. 11:1:1:1 for 8c – see ESI†), and the X-ray structure of 8c reveals the same configuration for all four elements of chirality (SCo*, SC*, Ra*, M*).
Ester substituted cyclopentadienyl complexes were readily prepared following in situ generation of sodium carbomethoxycyclopentadienide (Scheme 5, Table 2).22 As before, these complexes containing a methyl substituted stereogenic centre derived from 7a, 7c and 7d resulted in up to four solution stereoisomers.
 | ||
| Scheme 5 Diastereoselective synthesis of carbomethoxy substituted half-sandwich complexes 11. | ||
| Entry/diyne | R | Ar1 | Ar2 | Product/yielda (%) |
|---|---|---|---|---|
| a Isolated by column chromatography. | ||||
| 1 7a | Me | Ph | Ph | 11a 64 |
| 3 7c | Me | 4-CF3C6H4 | Ph | 11c 49 |
| 4 7d | Me | Ph | 4-CF3C6H4 | 11d 65 |
A non-racemic sample of 8a was synthesised starting with commercially available (S)-3-butyn-2-ol. Following ester formation with 5 as outlined in Scheme 2 (96%), followed by Sonogashira coupling with iodobenzene (>99%), (S)-7a was complexed by Method B to give (S)-8a in 74% yield. Chiral HPLC analysis gave a single peak in contrast to the two well separated peaks observed for racemic 8a. These results are consistent with the observation of four solution species of 8a by NMR spectroscopy at room temperature where interconversion between these species is rapid. Essentially no difference was observed in the 1H NMR of 8a recorded at 60 °C,23 and heating at higher temperatures resulted in decomposition. This is in marked contrast to 1 where heating at reflux in toluene results in phosphine dissociation and clean formation of (η5-cyclopentadienyl)(η4-tetraphenylcyclobutadiene)cobalt.8
In conclusion, we have demonstrated a short highly diastereoselective modular synthesis of new air-stable cobalt-based chiral-at-metal half-sandwich complexes obtained by oxidative cyclisation. The methodology is applicable to both substituted or unsubstituted cyclopentadienyl ligands and was readily adapted to the synthesis of a configurationally stable single enantiomer. These complexes provide an alternative to chiral metallocene frameworks as the basis of novel ligands, catalysts and materials.
The EPSRC (JA) is thanked for financial support. We also thank Caroline Taylor for some preliminary experiments, the EPSRC National Crystallography Service (Southampton) and the EPSRC National Mass Spectrometry Centre (Swansea).
Notes and references
- H. C. Malinakova, Chem.–Eur. J., 2004, 10, 2636 Search PubMed.
- (a) C. J. Richards and A. J. Locke, Tetrahedron: Asymmetry, 1998, 9, 2377 CrossRef CAS; (b) T. J. Colacot, Chem. Rev., 2003, 103, 3101 CrossRef CAS; (c) R. G. Arrayás, J. Adrio and J. C. Carretero, Angew. Chem., Int. Ed., 2006, 45, 7674 CrossRef.
- (a) H. Brunner, Angew. Chem., Int. Ed., 1999, 38, 1194 CrossRef CAS; (b) C. Ganter, Chem. Soc. Rev., 2003, 32, 130 RSC.
- (a) J. A. Gladysz and B. J. Boone, Angew. Chem., Int. Ed. Engl., 1997, 36, 550 CrossRef; (b) S. G. Davies, Aldrichimica Acta, 1990, 23, 31 CAS; (c) J. W. Faller, M. R. Mazzieri, J. T. Nguyen, J. Parr and M. Tokunaga, Pure Appl. Chem., 1994, 66, 1463 CAS.
- E. B. Bauer, Chem. Soc. Rev., 2012, 41, 3153 RSC.
- H. Brunner, Angew. Chem., Int. Ed. Engl., 1969, 8, 382 CrossRef CAS.
- (a) H. Brunner, G. Riepl, R. Benn and A. Rufińska, J. Organomet. Chem., 1983, 253, 93 CrossRef CAS; (b) Z. Zhou, C. Jablonski and J. Brisdon, Organometallics, 1993, 12, 3677 Search PubMed; (c) Y. Yu, C. Jablonski and J. Brisdon, Organometallics, 1997, 16, 1270 Search PubMed; (d) M. R. Meneghetti, M. Grellier, M. Pfeffer, J. Dupont and J. Fischer, Organometallics, 1999, 18, 5560 CrossRef CAS.
- (a) H. Yamazaki and Y. Wakatsuki, J. Organomet. Chem., 1977, 139, 157 CrossRef CAS; (b) H. V. Nguyen, M. R. Yeamine, J. Amin, M. Motevalli and C. J. Richards, J. Organomet. Chem., 2008, 693, 3668 CrossRef CAS.
- C. J. Taylor, M. Motevalli and C. J. Richards, Organometallics, 2006, 25, 2899 CrossRef CAS.
- Y. Wakatsuki and H. Yamazaki, Inorg. Synth., 1989, 26, 189 CAS.
- H. Brunner, Eur. J. Inorg. Chem., 2001, 905 CrossRef CAS.
- H. Brunner, K. Fisch, P. G. Jones and J. Salbeck, Angew. Chem., Int. Ed. Engl., 1989, 28, 1521 CrossRef.
- For extension of the Cahn-Ingold-Prelog system to polyhapto-ligands see: C. Lecomte, Y. Dusausoy, J. Protas, J. Tirouflet and A. Dormond, J. Organomet. Chem., 1974, 73, 67 Search PubMed , and also ref. 5.
- T. R. Ward, O. Schafer, C. Daul and P. Hofmann, Organometallics, 1997, 16, 3207 CrossRef CAS.
- An inversion barrier ΔG‡ for 9 of 56 kJ mol−1 has been estimated from NMR data where X = CH2, Y = +NMe2, R = H: I. O. Sutherland and M. V. J. Ramsay, Tetrahedron, 1965, 21, 3401 Search PubMed.
- S. L. Pira, T. W. Wallace and J. P. Graham, Org. Lett., 2009, 11, 1663 CrossRef CAS , and references therein.
- (a) S. E. Garner and A. G. Orpen, J. Chem. Soc., Dalton Trans., 1993, 533 RSC; (b) J. Polowin, S. C. Mackie and M. C. Baird, Organometallics, 1992, 11, 3724 CrossRef CAS.
- K. Mislow, Acc. Chem. Res., 1976, 9, 26 CrossRef CAS.
- H. Brunner, R. Oeschey and B. Nuber, Angew. Chem., Int. Ed. Engl., 1994, 33, 866 CrossRef.
- Relative to the Sc configuration the following torsions were observed: ω1 = 41.8°, ω2 = 53.4°, ω3 = 50.3°.
- J. W. Faller, J. Parr and A. R. Lavoie, New J. Chem., 2003, 27, 899 RSC.
- W. P. Hart, D. Shihua and M. D. Rausch, J. Organomet. Chem., 1985, 282, 111 CrossRef CAS.
- The energy barriers to interconverion of the isomers of 8a are estimated as ca. 60–80 kJ mol−1.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterisation data for 4c, 6, (S)-6, (S)-7a, 7c–g, (S)-8a, 8b–g and 11a, c and d. CCDC 889207 (8a) and 889208 (8c). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2cc34837c |
| This journal is © The Royal Society of Chemistry 2012 |