Molecular early main group metal hydrides: synthetic challenge, structures and applications†
Sjoerd
Harder‡
*
Stratingh Institute of Chemistry, University of Groningen, Nijenborgh 4, 9749 AG Groningen, The Netherlands. E-mail: s.harder@rug.nl; Tel: +31 50 3634322
First published on 26th September 2012
Abstract
Within the general area of early main group metal chemistry, the controlled synthesis of well-defined metal hydride complexes is a rapidly developing research field. As group 1 and 2 metal complexes are generally highly dynamic and lattice energies for their [MH]∞ and [MH2]∞ salts are high, the synthesis of well-defined soluble hydride complexes is an obvious challenge. Access to molecular early main group metal hydrides, however, is rewarding: these hydrocarbon-soluble metal hydrides are highly reactive, have found use in early main group metal catalysis and are potentially also valuable molecular model systems for polar metal hydrides as a hydrogen storage material. The article focusses specifically on alkali and alkaline-earth metal hydride complexes and discusses the synthetic challenge, molecular structures, reactivity and applications.
 Sjoerd Harder | Sjoerd Harder obtained his BSc, MSc and PhD degrees at the University of Utrecht in the Netherlands. After several post-doc stays at the University of Erlangen-Nürnberg (P. v. R. Schleyer), University of California at Berkeley (A. Streitwieser), University of Konstanz (H.-H. Brintzinger) he finalized his habilitation in 1998 at the University of Konstanz. He became an associate professor at the University of Duisburg-Essen (2004) and Chair in Inorganic Chemistry at the University of Groningen in his home country The Netherlands (2010). Within two years he accepted an offer at the University of Erlangen-Nürnberg and is currently moving back to Germany. |
1 Introduction
The element hydrogen holds a number of records: it is the first and lightest element in the periodic table, having the most simple atomic structure. Although it is hardly occurring in its elemental form on earth, it represents the most abundant element in the universe's chemical mass. Due to its high energy content it is currently in the picture as the energy carrier of the future.1Among its unique and peculiar properties is also the fact that it has an unusually high electronegativity for its position at the top of the group 1 elements (its Allred–Rochow's electronegativity of 2.2 is between those of B and C). As the 1s orbital has the correct energy for overlapping with orbitals of most other elements, hydrogen has the ability to combine with a plethora of chemical elements. As such, hydrogen acts like a chameleon and can have physical properties and reactivities varying from H+ and H-radical to H−. These various roles of the hydrogen ligand are especially underscored in its transition-metal chemistry which is exploited in a rich application in catalysis.2
The current feature article discusses hydrogen as a ligand in early main group metal complexes. Polar hydride compounds have found a rapidly increasing interest as storage materials for molecular hydrogen.3 Although previous reviews on early main group metal hydrides mainly deal with inorganic salt-like ion-pair compounds,4 this article focusses on molecular early main group metal hydride compounds and exclusively treats the group 1 and 2 metals. From a synthetic point of view, access to highly polar hydrides that are soluble in common organic solvents is challenging but also rewarding: this class of compounds fulfills an important role as intermediates in the rapidly developing field of early main group metal catalysis.5 This article will describe the historical development of this field, group 1 and group 2 separately treated, in a chronological order. Emphasis will be on complexes that have been unequivocally characterized by single crystal X-ray diffraction.
2 Saline early main group metal hydrides
Although these purely inorganic compounds are not part of the focus of this review, a short introduction on this class of salt-like compounds will help in putting the molecular main group hydrides in a better perspective. Group 1 and 2 metal hydrides form simple salt-like structures consisting of metal cations and hydride anions (Table 1).6,7,10| Group 1 MH | Δχa | M–Hb, Å | Lattice energyc, kJ mol−1 | Group 2 MH2 | Δχa | M–Hb, Å | Lattice energyc, kJ mol−1 |
|---|---|---|---|---|---|---|---|
| a Electronegativity differences based on Allred–Rochow values. b Average experimental values (ref. 6), except for BeH2 (ref. 10). c Calculated values taken from ref. 7. | |||||||
| LiH | 1.2 | 2.04 | 858 | BeH2 | 0.7 | 1.41 | 3205 |
| NaH | 1.2 | 2.45 | 782 | MgH2 | 1.0 | 1.95 | 2791 |
| KH | 1.3 | 2.86 | 699 | CaH2 | 1.2 | 2.32 | 2410 |
| RbH | 1.3 | 3.03 | 674 | SrH2 | 1.2 | 2.49 | 2250 |
| CsH | 1.3 | 3.20 | 648 | BaH2 | 1.2 | 2.67 | 2171 |
All group 1 metal hydrides are of the NaCl structural type in which each M+ cation is surrounded by six H− anions and vice versa each H− anion by six M+ cations. In contrast, the series of group 2 metal hydrides possesses different structures. The heavier metal hydrides MH2 (M = Ca, Sr, Ba) have at room temperature a PbCl2-type structure (cotunnite) in which the metal cations have the high coordination number of nine, whereas H− anions have short contacts to four or five metal cations. The room temperature form of MgH2 has a TiO2-type structure (rutile) in which Mg2+ binds to six H− anions and each H− anion to three Mg2+ cations. The structure of its lightest group member, BeH2, has been shrouded in mystery for a long time (in fact, even its existence has been subject of discussion).8 The originally proposed one-dimensionally polymeric structure of [BeH2]∞, a model often found in student manuals, consists of a linear array of tetrahedral BeH42− ions connected by Be2+ cations, i.e. edge-sharing BeH4 tetrahedra (1).9 More recent accurate structural studies show a three-dimensional polymeric network of corner sharing BeH4 tetrahedra in which each Be is four-coordinate and each hydride ligand connects two Be centers (2).10
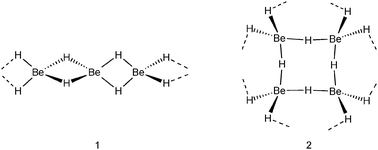
The nature of the metal–hydride bond is naturally dependent on electronegativity differences which for group 1 metal hydrides hardly vary (Δχ = 1.2–1.3) but point to highly ionic character. It should, however, be noticed that also cation size plays a dominant role: the smaller cations are able to effectively polarize only weakly bound 1s2 electrons of the H− ion. The free H− ion is estimated to have an ionic radius of 2.08 Å but is highly compressed in early main group salt structures (ionic radii of 1–1.4 Å).6 Compressibility increases along the row Cs < Li and is partially due to increased covalent character. Especially in BeH2 (Δχ = 0.7) covalent contributions can be high and this was the reason to propose the wrong linear structure (1) which was explained by 3-center-2-electron bridging of H− ligands (vide supra). The heavier group 2 hydride complexes are largely ionic in nature, however, some covalent character also has been found for MgH2.11
Noteworthy are also the very high lattice energies for early main group metal hydrides (Table 1). These decrease along group 1 from Li to Cs and greatly determine their chemical reactivity. Whereas NaH–CsH salts are increasingly reactive towards moisture, the salt with the highest lattice energy, [LiH]∞, is essentially inert. Below red heat temperatures it is unaffected by O2 or HCl.12 Neither does it react with tertiary alcohols at room temperature.13 Group 2 metal hydrides show even larger lattice energies. The high lattice energy for MgH2 seriously affects its practical use as a promising hydrogen storage material and is the reason why high temperatures of >300 °C need to be applied for the redox reaction: MgH2(s) → Mg0(s) + H2(g).14 Commercially available CaH2 reacts only with water but not with other common organic functional groups (halides, ketones, cyanides). This makes it the drying agent of choice for a large array of organic solvents.
The reactivity of such highly stable saline metal hydrides can be strongly increased by taking them out of their crystal lattice and bringing them into a soluble form. Commercially available solutions of LiH/BH3 and LiH/AlH3 are probably the best sold reduction reagents,15 however, these should rather be viewed as ‘ate’ complexes Li+BH4− and Li+AlH4− in which the hydride ligands are strongly bound to the main group 3 element and Li+ only acts as a charge balancer. Although these compounds show polar hydride reactivity, their nature and structures are only remotely related to saline group 1 and 2 hydrides and therefore not part of this review.
The high lattice energies encountered also make it a real challenge to synthesize early main group metal hydride complexes. Whereas for most organolithium compounds large varieties of adducts have been structurally characterized, e.g. (PhLi·Et2O)4, (PhLi·TMEDA)2 or (PhLi·PMDTA)2, hardly any hydride complexes are known. For the alkali metals, complexes of the form L0–M+–H− (in which L0 is a neutral ligand) should be feasible. For the alkaline-earth metals complexes of the type L0–M2+(H−)2 or L−–M+–H− could exist. However, facile ligand exchange reactions that are typical in early main group chemistry and high lattice energies of the insoluble salts [MH]∞ and [MH2]∞ make the isolation of such ligand-stabilized compounds very challenging. Although the smaller metal cations display the largest lattice energies, metal hydride complexes with larger metal cations are the most difficult to obtain. This is due to the rapidly decreasing M–H bond strengths and therefore faster ligand exchange processes in going down the groups. Also, the large coordination sphere of these heavier metal cations gives rise to intermolecular interaction and easier ligand exchange. However, with the right combination of ligands soluble early main group metal hydride complexes can be obtained. As high lattice energies of these saline metal hydrides do not allow for synthesis of their complexes starting from the metal hydride salt itself, their synthetic protocol is usually based on the in situ formation of a molecular metal hydride in the presence of suitable stabilizing ligands.
3 Mixed-metal hydride complexes
A variety of transition metal complexes with incorporated group 1 or 2 metal hydrides have been reported.16–24 On account of the high electropositivity of early transition metals and lanthanides, their mixed-metal complexes can often be described as aggregates containing a hydride ligand.Floriani et al. reported a ZrIV complex that can act as a “carrier for NaH or KH in toluene”.16,17 Two disc-like molecules consisting of a fourfold deprotonated porphyrinogen and ZrIV sandwich a (MH)2 dimer (M = Na, K) (3). Although such compounds may react hydridic and thus could be seen as a hydrocarbon-soluble form of an alkali metal hydride, it would be more appropriate to describe these as zirconate complexes (LZrH−) that are bridged by two M+ ions. This vision is mainly based on estimated bond strengths: the Zr–H bond is not only more covalent, and therefore stronger than the M–H bonds, but also has an electrostatic component that is based on attraction to a highly Lewis-acidic ZrIV center (instead of to the much weaker M+ Lewis acid). Similar ambiguities can be found in for example (Cp*2ZrH2)(LiH).18 This complex crystallizes as a trimer (4) which can be described either as three Cp*2ZrH2 molecules trapping a cyclic LiH-trimer or as three anionic zirconates held together by three Li+ ions: [(Cp*2ZrH3−)(Li+)]3. The report correctly describes this complex as a zirconate.18
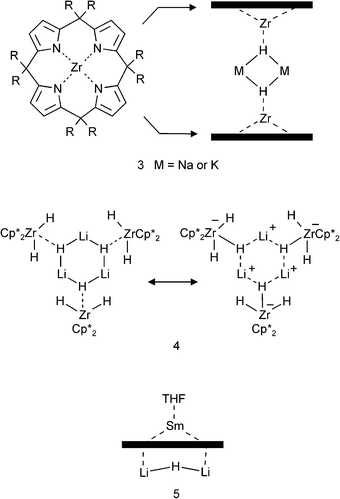
Several more examples of such cases can be found in Andersen's (Cp*2TiIIIH2−)(Li+·TMEDA) and Mach's (Cp*2TiIIIH2−)2(Mg2+),19 Piers's [(ArN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ScCl·THF)3H](Li+),20 Hou's sphere-like complex Cp*6H15K3SmIII6THF6,21 Nöth's [(tBuCH2O)5Al3H5−](Li+)22 or Robinson's self-assembled organometallic sphere that resembles the seams of a tennis ball and “entraps” NaH.23 In all these cases, the hydride anions are strongly bound to highly charged cations and consequently should be assigned to them. These species should be described preferably as containing an ‘ate’ anion.
ScCl·THF)3H](Li+),20 Hou's sphere-like complex Cp*6H15K3SmIII6THF6,21 Nöth's [(tBuCH2O)5Al3H5−](Li+)22 or Robinson's self-assembled organometallic sphere that resembles the seams of a tennis ball and “entraps” NaH.23 In all these cases, the hydride anions are strongly bound to highly charged cations and consequently should be assigned to them. These species should be described preferably as containing an ‘ate’ anion.
Some exceptions or borderline compounds may be found in following examples. Gambarotta's samarium(III) complex consisting of a fourfold deprotonated porphyrinogen (like in 3), SmIII and a Li2H+ ion coordinated to the opposite site of the macrocycle (5, the “through-ring” Sm–H bond distance of 2.25(7) Å is rather long).24 Also Veith recently reported that Li–Al hydride represents an example in which LiH could be described as being trapped in an organometallic complex (Fig. 1).25 Although the complex has suggestively been reported as a cubane-like (LiH)4 tetramer that is stabilized by interactions with three (Me3Si)2NAlH molecules, it would be better to describe this species as three aluminate anions (Me3Si)2NAlH2− that are bridged by Li+ connectors. The remaining Li+H− unit in the middle (the diagonal of the Li4H4 cube), however, can be regarded as a truly encapsulated LiH unit (there are no contacts of this particular H− ion to AlIII).
3(LiH) by Veith et al.;25 carbon atoms on Me3Si are not shown for clarity.](/image/article/2012/CC/c2cc33478j/c2cc33478j-f1.gif) | ||
| Fig. 1 The crystal structure of [(Me3Si)2NAlH2−](Li+)3(LiH) by Veith et al.;25 carbon atoms on Me3Si are not shown for clarity. | ||
4 Group 1 metal hydride complexes
Very active forms of LiH can be obtained simply by deprotonation of H2 by tBuLi but this process needs forcing conditions (200 bar).26 Polar (co)solvents like TMEDA or THF drastically lower the energy barrier and allow conversion of nBuLi into LiH even at atmospheric pressures.27 This method has been extended by Brandsma et al. to the heavier group 1 metals. Deprotonation of H2 with “superbase mixtures” gave highly active, but not necessarily soluble forms of NaH, KH and even CsH (eqn (1)).27–30 | (1) |
 | (2) |
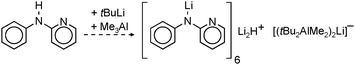 | (3) |
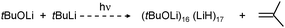 | (4) |
In general, the in situ prepared alkali metal hydride species precipitate from solution in their cubic crystal lattices. However, these remaining suspensions are extremely finely dispersed and highly reactive (the presence of nanoparticles should not be excluded, vide infra). The precipitation of these particles nicely demonstrates the challenge of finding solubilizing ligands that prevent conversion of molecular metal hydrides into highly stable crystalline salts. Some well-established examples of metal hydride incorporation into organometallic complexes or clusters are described below.
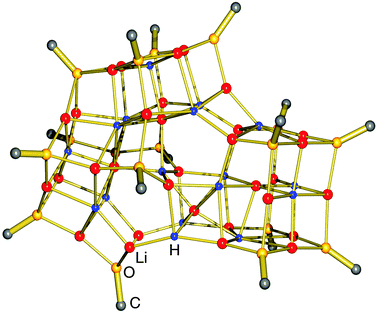
First well-defined lithium complexes with interstitial hydride have been found by Wheatley et al.33 who demonstrated the favourable stability of clusters in which the H− ion resides in an octahedral hole spanned by 6 Li+ ions (Fig. 2).34–36 These clusters are prepared by a standard route in which β-H elimination from tBuLi is held responsible for in situ formation of H− (eqn (3)). It is not easy to prove the presence of the light hydride ion in a Li6H− unit with standard X-ray structural methods. The hydride anion also often remains NMR silent. However, a recent neutron diffraction study resolved the ambiguity presented by these species.36 Considering the high molecular weight of the total cluster (1072), the lithium hydride content is rather low.
![The cationic cluster {[(2-pyridyl)NPh]Li8H}+ by Wheatley et al.33 (Ph groups only partly shown for clarity); slight deviation from projection along an approximate threefold axis.](/image/article/2012/CC/c2cc33478j/c2cc33478j-f2.gif) | ||
| Fig. 2 The cationic cluster {[(2-pyridyl)NPh]Li8H}+ by Wheatley et al.33 (Ph groups only partly shown for clarity); slight deviation from projection along an approximate threefold axis. | ||
A most spectacular example of high lithium hydride incorporation can be found in the cluster (tBuOLi)16(LiH)17 which was characterized using X-ray diffraction by Thomas et al. (Fig. 3).37 This gigantic cluster has been prepared by in situ generation of LiH through photostimulated β-H elimination of tBuLi in the presence of the bulky lithium alkoxide tBuOLi (eqn (4)). Crystals were obtained after keeping a pentane solution for several weeks at −90 °C. Although yields are not given, the procedure is reported as being reproducible.
 | ||
| Fig. 3 Crystal structure of (tBuOLi)16(LiH)17 by Thomas et al.37 (tBuO groups only partly shown for clarity); the crystallographic mirror plane is close to the plane of projection. | ||
The crystal structure shows a rather complicated aggregate with crystallographic mirror-symmetry (Fig. 3). Although it is difficult to describe its structure, numerous distinctive cubic features, typical for group 1 metal salts, can be recognized easily. The aggregate can be thought to consist of three fused partial cubic units with tBuO on the corners and Li+ and H− ions filling the inner positions; such a tentative complete cubic unit would be of composition (tBuO)8Li13H6. The formation of [LiH]∞ is apparently disrupted by the incorporation of the sterically demanding tBuO groups.
Very recently, Stasch could obtain a simpler more defined LiH-containing complex.38 The LiH is formed here in situ from nBuLi and PhSiH3 similar to the reaction discussed for KH formation (eqn (2)). Crystals can be obtained in a reproducible yield of 62% and were characterized by X-ray diffraction (Fig. 4). The hydride ions have been located and Li–H distances vary from 1.96(4)–1.99(4) Å (which is close to the 2.04 Å value in cubic LiH). The 1H NMR of a benzene solution shows one broad resonance for all hydride ligands. Above 60 °C, the complex is quite unstable and loses the encapsulated (LiH)4 by precipitation of saline lithium hydride.
![Crystal structure of (LiH)4 sandwiched between two [Ph2P(Ph)NLi] dimers by Stasch38 (only the Cipso atoms of the Ph groups are shown).](/image/article/2012/CC/c2cc33478j/c2cc33478j-f4.gif) | ||
| Fig. 4 Crystal structure of (LiH)4 sandwiched between two [Ph2P(Ph)NLi] dimers by Stasch38 (only the Cipso atoms of the Ph groups are shown). | ||
5 Group 2 metal hydride complexes
Early work focusses on the syntheses of soluble beryllium hydride complexes of the group 2 metals.39–46 Although BeH2 has the highest lattice energy, Be2+ being the smallest cation also has the smallest coordination sphere and is easy to saturate with small ligands like hydrides. Moreover, the considerable metal–ligand bond strengths prevent ligand exchange and thus formation of insoluble BeH2.Early reports, mainly by G. E. Coates et al., describe the synthesis of hydrocarbon-soluble beryllium hydride compounds. Solutions of RBeH (R = Me, Et, tBu, nPe, Ph) could be obtained by mixing R2Be, BeCl2 and LiH in boiling ether.39–41 A more efficient method for in situ generation of hydrides is based on ligand exchange with Me3SnH which after reactions with R2Be (R = Me, Et) gives RBeH that has been isolated as amine adducts.42,43 An elegant method for hydride formation is β-hydride elimination: iPr2Be thermally decomposes to iPrBeH and H2CCMe2.44 It should be mentioned that the characterization of these compounds is limited to methods available in the 60–70's. An early crystal structure of a beryllium hydride was reported for the beryllate (Et2BeH−)Na+·Et2O that crystallized as a dimer with H ligands bridging the Be centers (Be–H = 1.49 Å) but also with short Na–H contacts of 2.40 Å.45 The first crystal structure of a “true” beryllium hydride complex was reported already as early as 1983 by Coates et al.: [Me2NCH2CH2N(Me)BeH] (Fig. 5a).46 Interestingly, the amide anions form the bridges between the Be centers whereas the H− ligands reside at a terminal position (Be–H = 1.38(1) Å). Parkin et al. reported the crystal structure of the scorpionate complex TptBuBeH which features a Be–H distance of 1.23(7) Å (Fig. 5b).47 As the lattice energy for [BeH2]∞ is enormous (Table 1), this complex is likely thermodynamically less stable. However, the strength of the Be–H bond, which is expected to have considerable covalent character, precludes any ligand exchange via Schlenk equilibria. Moreover, the very condensed coordination sphere of Be would not allow for the formation of the coordination complex (TptBu)2Be. The complex TptBuBeH is well-soluble in benzene and the hydride ligand shows a singlet 1H NMR resonance at 5.00 ppm.
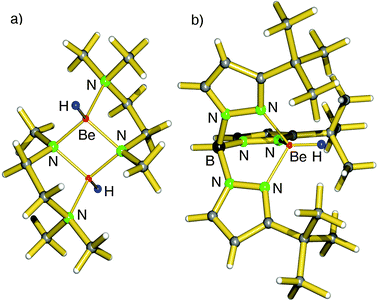 | ||
| Fig. 5 (a) Crystal structure of the first beryllium hydride complex by Coates et al.46 (b) Crystal structure of TptBuBeH by Parkin et al.47 | ||
Apart from beryllium hydride complexes, also the magnesium hydride chemistry was developed at an early stage. Although the existence of compounds like RMgH has been the subject of interest and controversy,48 extensive work by Ashby et al. describes the syntheses of a large variety of magnesium hydride complexes. Their synthetic method is based on ligand exchange between R2Mg and ether-soluble LiAlH4. Although this protocol seems troublesome and outcomes might be unsure (two different anions are mixed with three different metal cations) the following compounds are claimed on the basis of elemental analysis and IR studies: PhMgH, PhMg2H3, MeMg2H3 and CpMgH.49,50 Additional NMR and MW determination studies on CpMgH·THF led us to propose a dimeric structure with bridging H− ions and terminal Cp− and THF ligands.51 Also complexes of the type XMgH, in which X = Cl, Br, OR and NR2, have been subject of intensive research but no clear-cut structural characterization has been presented.52–55
Access to soluble forms of calcium hydride has been more troublesome, probably because organocalcium chemistry only started to develop since the last two decades.5,56–60 Therefore, first examples describe synthetic routes based on highly active Ca vapour. Mixed with organics like benzene, these produced PhCaH species through C–H activation.61,62 The compounds have been characterized by quench experiments but, as for the early magnesium hydrides, no NMR or structural proof is available.
First definite proof for a well-defined magnesium hydride complex was found in the form of an “inverse crown ether”, i.e. the hydride anions are encapsulated in a ring of alternating amide and metal ions (Fig. 6).63,64 The synthesis is based on the thermal decomposition of the magnesate complexes [(iPr2N)3Mg−] M+ (M+ = Na+, K+) in which a hydride anion is generated in situ by β-hydride elimination in the iPr2N− anion (eqn (5)). The Mg–H distances in the resulting Na/Mg and K/Mg complexes are 1.89(2) and 1.94(2) Å, respectively, and both show similar 1H NMR resonances at 3.76 and 3.78 ppm, respectively.
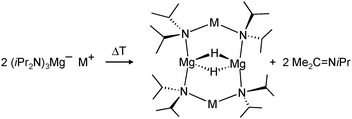 | (5) |
 | (6) |
 | (7) |
 | (8) |
 | (9) |
Synthesis of well-defined alkaline-earth metal hydride complexes of the heavier group 2 metals (Ca, Sr, Ba), which would be highly dynamic, is expected to be more challenging. First attempts to synthesize such complexes by Hanusa et al. were based on borane extraction from a calcium boron hydride precursor: (Me3Si)3CpCa(HBEt3), however, no transfer of the hydride from B to Ca was observed.65 Similarly, H. W. Roesky et al. reported that this approach also fails for magnesium or strontium hydride complexes.66,67
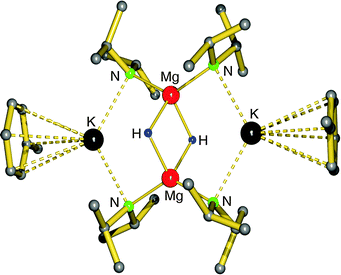 | ||
| Fig. 6 Crystal structure of the “inverse crown ether” complex encapsulating two hydride anions by Mulvey et al.63,64 | ||
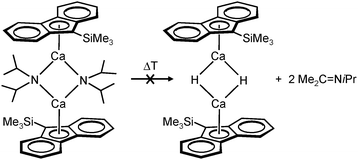
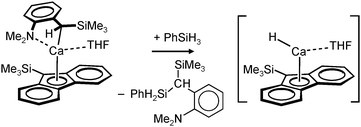
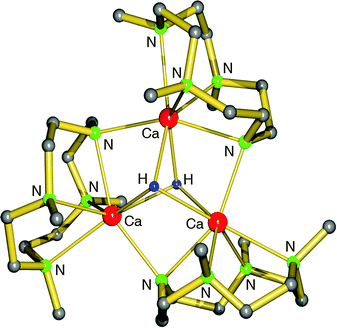
Harder et al. set out to prepare first defined calcium hydrides.68 The rationale behind their approach was to make L–Ca–R complexes in which L is a bulky ligand and R a reactive group that could be converted in situ into a hydride ligand. As KN(iPr)2 had previously been shown to be prone to thermal decomposition into KH and Me2C=N−iPr,63,64 (9-Me3Si-fluorenyl)CaNiPr2, which crystallizes as a dimer, was heated in benzene solution (eqn (6)) but insignificant decomposition was observed. In a second approach, a similar but monomeric benzylic calcium compound was treated with PhSiH3 (eqn (7)). Although formation of benzyl-SiH2Ph proves exchange of the benzyl anion for H−, only the homoleptic calcium complex with two fluorenyl ligands could be isolated, implying that the Schlenk equilibrium is operative here. As strongly coordinating multidentate ligands could stabilize a heteroleptic calcium hydride, it seemed that the bulky scorpionate ligand TptBu would be a good candidate to prevent formation of (TptBu)2Ca. The observation that attempted synthesis of homoleptic (TptBu)2Ca was not possible even under extreme conditions69 strengthened this idea. Reaction of TptBuCaN(SiMe3)2 with PhSiH3, however, gave the homoleptic complex (TptBu)2Ca.68 The crystal structure gave insight into how two bulky scorpionate ligands can coordinate to a single Ca2+ center: one of the ligands showed the normal tridentate coordination mode (three Ns bind the metal) whereas the other is only bound bidentate (two Ns bind the metal) and displays an additional B–H⋯Ca2+ interaction, i.e. the “sting of the scorpion”.
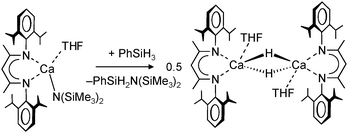
Use of the bulky β-diketiminate ligand DIPP-nacnac, (2,6-iPr2C6H3)NC(Me)C(H)C(Me)N(2,6-iPr2C6H3), finally led to a stable heteroleptic calcium hydride. Reaction of (DIPP-nacnac)CaN(SiMe3)2·THF with phenylsilane in hexane (eqn (8)) gave after cooling a batch of large colourless crystals of the heteroleptic dimer: [(DIPPnacnac)CaH·(THF)] (Fig. 7). The crystal structure shows bridging H− ligands which could be located (average Ca–H distance: 2.15(3) Å). The 1H NMR spectrum shows for the hydride ligand a relatively sharp singlet at 4.45 ppm. The hydride complex dissolves well in benzene and shows a remarkable stability. Forcing reflux conditions do only after prolonged reaction times lead to ligand exchange into (DIPPnacnac)2Ca and insoluble [CaH2]∞. This is rather surprising in the light of the fact that homoleptic (DIPPnacnac)2Ca is highly stable on account of intramolecular C–H⋯π bonding.70
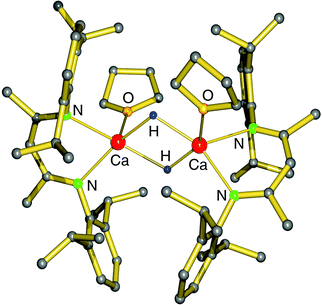 | ||
| Fig. 7 Crystal structure of the first calcium hydride complex by Harder et al.68 | ||
Using the same DIPPnacnac ligand and a similar synthetic protocol, DIPPnacnacMgnBu + PhSiH3 → DIPPnacnacMgH + nBuSiH2Ph, Jones et al. could isolate an analogue dimeric magnesium hydride. As this particular synthesis was run in the absence of polar THF, a solvent-free dimer [DIPPnacnacMgH]2 with bridging H− ligands crystallized.71 Crystallization in the presence of THF, however, led to the THF-adduct [DIPPnacnacMgH·THF] which has a similar structure to that of the calcium hydride shown in Fig. 7. Coordination of THF hardly affects the Mg–H bond distances which are 1.96(3) and 1.95(2) Å in the THF-free complex and the THF-adduct, respectively. Instead, THF-coordination elongated the Mg–N bonds by circa 0.1 Å. Also the 1H NMR shifts are similar: 4.03 and 4.21 ppm, respectively.
Harder et al. found an efficient alternative for the synthesis of [DIPPnacnacMgH]2: overnight thermal decomposition of DIPPnacnacMgNH(iPr)BH3 gave the product in 89% isolated yield.72 This route represents a good alternative to the silane route which for Mg is slow and low-yielding (2 days; 40%).
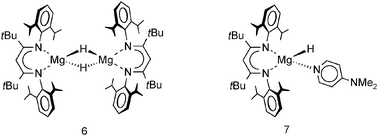
In later work, Jones et al. reported a dimeric magnesium hydride complex with bulky tBu substituents in the backbone (6).73 Although these remote substituents induce more steric bulk at the metal center by “pushing” DIPP groups towards the metal, the Mg–H distances in this complex are shorter, i.e. 1.84(5) Å compared to 1.96(3) Å, but within standard deviation; the reported 1H NMR shift is 3.83 ppm. The shortest Mg–H distance of 1.75(7) Å was found in a monomeric magnesium hydride complex (7) which was obtained by addition of DMAP to a solution of the dimer; the reported 1H NMR shift is 4.65 ppm. Hitherto, this is the only example of a complex with a terminal Mg–H bond.
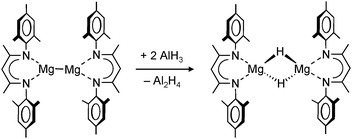
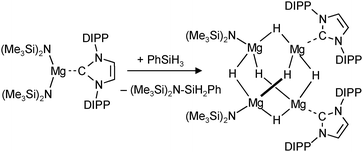
A unique redox reaction between a dimeric MgI complex and AlH3 (in the presence of an N-heterocyclic carbene) yielded a somewhat less sterically shielded magnesium hydride dimer (eqn (9)) with mesityl substituents in the nacnac ligand (Mg–H = 1.88(2) Å).74 Although a crystal structure could be obtained, the species is less defined in solution and decomposes through ligand exchange in the homoleptic species. This nicely illustrates how a slight decrease in the steric bulk of the supporting ligand results in considerable loss of stability. This more open magnesium hydride is also highly reactive with silicone grease and a dimer with bridging H− and Me2HSiO− ions could be characterized74 (in addition, a dimeric complex with H− and I− ions was found).73
 | (10) |
 | (11) |
 | (12) |
 | (13) |
 | (14) |
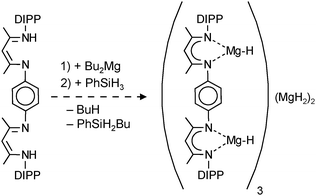
Earlier work by Michalczyk describes the synthesis of MgH2 adducts with bidentate ligands by reaction of nBu2Mg with PhSiH3 in the presence of DME or TMEDA. This route, however, led to insoluble, amorphous powders.75 Hill et al. attempted to stabilize the MgH2 unit with a strongly coordinating, bulky N-heterocyclic carbene. The in situ preparation of MgH2 from a Mg amide precursor was found to be incomplete and they isolated a mixed amide/hydride cluster (eqn (10)).76 The cluster can be described as a C2-symmetric hexanuclear compound with an adamantane-like Mg4H6-core and terminal amide and carbene ligands (Fig. 8). The Mg–H bonds range from 1.86(1)–1.90(1) Å and the 1H NMR shows one broad signal at 2.52 ppm for the hydride ligands, indicating exchange on the NMR time scale. The complex is described as being robust and so far defies further exchange of amide ligands for hydride anions.
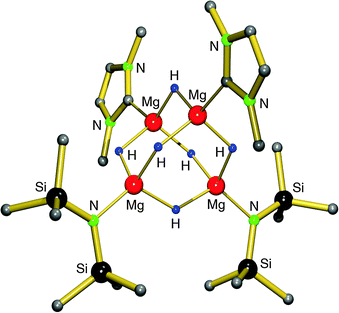 | ||
| Fig. 8 Crystal structure of a tetranuclear magnesium hydride/amide complex by Hill et al.;76 the DIPP substituents at the N-heterocyclic carbene ligand are only partially shown for clarity. | ||
Based on the successful use of β-diketiminate (nacnac) ligands in the stabilization of group 2 metal hydride clusters, Harder et al. prepared a tetranuclear magnesium hydride cluster by using a coupled nacnac ligand (eqn (11)).77 The Mg4H4-core of the structure (Fig. 9a) shows a similar adamantane-like geometry as Hill's tetranuclear magnesium hydride cluster with Mg–H bond distances varying from 1.77(1) to 1.85(1) Å. The Mg2+ ions are situated at the corners of a hypothetical tetrahedron of which four vertices are bridged by hydride ligands and two opposite vertices by the bis-nacnac ligands. Interestingly, there are two different hydride positions, H1 and H2, which give two triplets in the 1H NMR spectrum at low temperature (2.85 and 3.64 ppm, 2JH–H = 8.5 Hz). At higher temperature these signals coalesce into a singlet (Tcoal = 25 °C, ΔG‡ = 56(1) kJ mol−1).
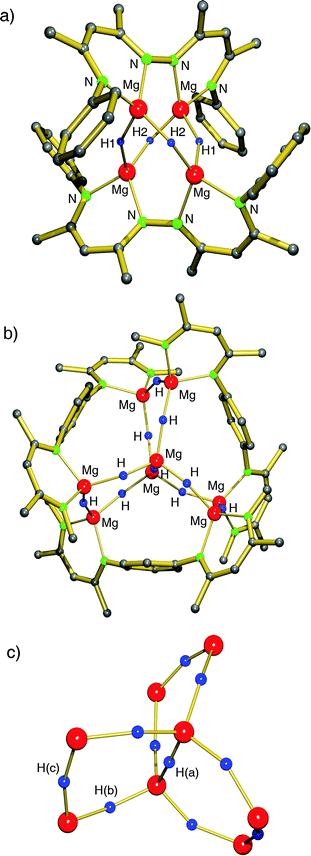 | ||
| Fig. 9 Crystal structure of bis(nacnac)-stabilized magnesium hydride clusters by Harder et al.; partially shown for clarity.77,78 | ||
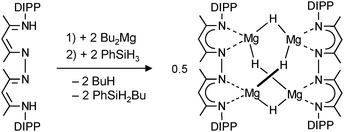
By using bis-nacnac ligands with a larger para-phenylene bridge (eqn (12)), Harder et al. could obtain a significantly extended magnesium hydride cluster in a reproducible yield of circa 50%.78 The larger span of the bridge allows for incorporation of two MgH2 units (these are likely formed in situ by the reaction of an excess of nBu2Mg with PhSiH3). The crystal structure of this octanuclear magnesium hydride complex, containing a total of ten hydride ligands, is shown in Fig. 9b; Mg–H bond distances range from 1.72(3) to 1.90(3) Å (average 1.81(3) Å). Although the cluster is crystallographically C2-symmetric, the propeller-like orientation of the ligands induces near C3-symmetry. The Mg–H framework (Fig. 9c) is similar to a three-spoked wagon wheel in which three different kinds of hydride positions are present: (a) at the axis, (b) on the spokes, and (c) at the outside. These three different hydrides can be observed in 1H NMR as a heptet (H(a), 0.55 ppm, 2JH–H = 4.5 Hz), double doublet (H(b), 1.72 ppm, 2JH–H = 4.5 and 5.2 Hz) and a triplet (H(c), 2.60 ppm, 2JH–H = 5.2 Hz), respectively. These coupling patterns can not only be observed in solutions in aromatic solvents but also in THF. Moreover, the three signals with H–H coupling can even be observed at 100 °C. This is not only strong evidence for preservation of the solid state structure in solution but also demonstrates its unusual static behaviour.
The observed hydride–hydride coupling represents the first examples of He–Mg–H magnetic coupling in magnesium hydride species. These multinuclear clusters serve as molecular model systems for bulk MgH2 as a hydrogen storage material (vide infra).
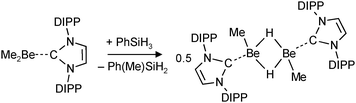
Very recent work on group 2 metal hydride chemistry relates to the isolation and crystallographic characterization of MeBeH stabilized by a bulky N-heterocyclic carbene (NHC).79 This complex was obtained by the “silane method” (eqn (13)), a procedure that proved to be highly successful for isolation of calcium and magnesium hydride complexes.68,71,73,76–78 In contrast to the earlier discussed [Me2NCH2CH2N(Me)BeH]2 (Fig. 5a), in dimeric [MeBeHNHC]2 the hydride ligands are bridging with Be–H distances of 1.55(2) Å (the 1H NMR shift is 2.33 ppm). Attempted isolation of NHC stabilized BeH2 by further reaction of the product with PhSiH3 led to ring opening of the carbene ligand.
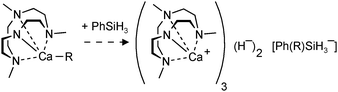
Hitherto, no examples of well-defined strontium and barium hydride complexes have been reported. As pointed out earlier, challenges to isolate and stabilize group 2 metal hydride complexes rapidly increase with metal size. The latest work on group 2 metal hydride chemistry is a report by Okuda et al. on the second example of a calcium hydride complex.80 Using the “silane method” a calcium hydride complex was obtained that is stabilized by a tetradentate cyclene-derived monoanionic ligand (eqn (14)). The complex crystallizes as a trimer but one of the hydride ligands is lost to the silane byproduct, forming a non-coordinating hypervalent silicate (Fig. 10). The trimeric nature of this complex is likely due to the fact that the cyclene anion is a less bulky ligand than DIPPnacnac− and the amide is an excellent bridging group. The trimeric bonding scheme allows for bridging amide Ns but only leaves place for two bridging H− ions at the two faces of the Ca3 triangle. The third H− ion is incorporated in the silicate which should be considered an intermediate on the pathway for formation of calcium hydrides (see eqn (2)). Three different complexes, all with the same trimeric calcium hydride cation but with different silicate anions, have been isolated. The Ca–H bond distances range from 2.03(8) to 2.50(9) Å (average: 2.33(9) Å); their 1H NMR chemical shift is found at 3.99 ppm.
 | ||
| Fig. 10 Crystal structure of a cationic trimeric calcium hydride complex stabilized by tetradentate ligands (Okuda et al.);80 the non-coordinating anion is either Ph3SiH2−, Me3SiN-SiPh3− or Ph(H)C-SiHPh2−. | ||
6 Relevance and applications in the field
Apart from being fundamentally important, early main group metal hydride complexes are also highly relevant species in various fields, e.g. in chemical reduction, as versatile synthons in organometallic chemistry, in catalysis or in hydrogen storage.Early work on hydrocarbon-soluble magnesium hydrides like X–Mg–H (in which X is a carbanion, alkoxide or amide) was motivated by application in selective reduction. Highly bulky amides like [Me3Si(tBu)N] MgH show excellent stereoselectivities in the addition to cyclic ketones.81 Apart from selectivities also activities of these reagents are clearly boosted by their good solubility.
Similar observations have been made by Harder et al. who investigated the reactivities of the first soluble calcium hydride complex [DIPPnacnacCaHTHF]2 (Fig. 7). Whereas solid CaH2 hardly reacts with any organic functional group and is primarily used as a drying agent for organic solvents containing a variety of functional groups, the solubilized calcium hydride displays smooth reactivity to a large array of substrates (Scheme 1) and gives access to a series of new calciumorganyl complexes mostly in crystalline purity.82 For this reason [DIPPnacnacCaH·THF]2 has been nicknamed “worker-bee” in calcium chemistry.
 | ||
| Scheme 1 Reactions of the Ca–H functionality (for simplicity represented as a monomer). | ||
One of the motivations for the syntheses and investigation of molecular group 2 metal hydrides originates from the proposal by Harder et al. that such complexes could be key intermediates in catalytic reactions like alkene hydrosilylation (Scheme 2, cycle A).31 Each step in the cycle has been separately proven in a stoichiometric protocol. The fact that for alkene hydrosilylation homoleptic R2Ca complexes are more efficient catalysts than heteroleptic LCaR species suggests that also solubilized (CaH2)n aggregates could be active catalysts. Indeed, experimental observations hint the possible existence of larger hydride-rich clusters which are stabilized by ligands in the outer-shell (Scheme 2, route B).31 It is likely that undefined nanoclusters are formed prior to precipitation of [CaH2]∞. Although hydride complexes can also be used as catalysts in ketone hydrosilylation, it is unlikely that the metal hydride complex is the “true” catalyst in this process.83
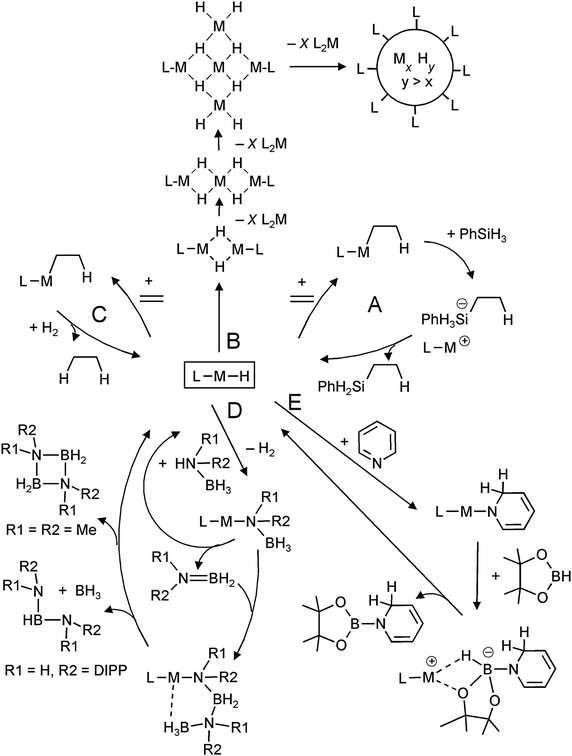 | ||
| Scheme 2 Metal hydride based intermediates in catalysis. | ||
The existence of group 2 metal hydride complexes led to the question whether such species could be used in hydrogenation catalysis (Scheme 2, cycle C). Harder et al. could show that [DIPPnacnacCaH·THF]2 (Fig. 7) is catalytically active in alkene hydrogenation with molecular H2 (20 bar).84 Also in situ generated calcium, strontium and potassium hydride species were found to be active catalysts. These represent the first non-transition metal catalysts for alkene hydrogenation.
Groups of Harder, Hill and Jones have shown that molecular alkaline-earth metal hydride complexes are excellent precursors in the syntheses of metal amidoborane complexes.72,85–90 These species have found renewed interest as hydrogen storage materials.91 Harder et al. found that the DIPPnacnac ligand used to stabilize molecular hydrides also serves as an excellent template for the stabilization of the thermal decomposition products of group 2 metal amidoborane complexes.72,85,86,88 It has been proposed that molecular metal hydride complexes are important intermediates in the elimination of hydrogen upon thermal decomposition of these metal amidoboranes.
Magnesium hydride complexes are also catalytic intermediates for condensation of ammonia–borane derivatives (Scheme 2, cycle D). Substituents on N determine the course of this reaction. Harder et al. introduced a catalytic route for the synthesis of HB[NH(DIPP)]2 from (DIPP)NH2BH3,92 whereas Hill et al. found that two smaller Me substituents on N gave the ring-like product (Me2NBH2)2.87 Similarly, heavier metal hydrides like solvated SrH2 complexes have been proposed as intermediates in such reactions but hitherto there is no precedence for well-defined complexes.89
Hill et al. observed that Jones's molecular magnesium hydride, [DIPPnacnacMgH]2, dearomatizes pyridine by a 1,2-addition (which later isomerizes to the 1,4-addition product),93 a reaction that is well-established for group 1 metal hydrides.94 This is the basis for catalytic dearomatization–hydroboration of pyridine derivatives (Scheme 2, route E).95 There are indications that the magnesium hydride is a recurrent species in this cycle. This strongly contrasts with the earlier reported Ca-mediated hydroboration of alkenes in which the calcium hydridoborate complex is the most stable species and disproportionation into a hydride and borane seems unlikely.96 The magnesium-hydride mediated catalytic hydroboration has very recently been extended to addition–hydroboration of aldehydes and ketones.97
Solid saline [MgH2]∞ is currently receiving tremendous attention as a hydrogen storage material. Although this material is not very rich in hydrogen (7.7 wt%), it has the main advantage of reversibly releasing or taking up hydrogen: MgH2 ⇆ Mg + H2.98 However, its relatively high thermodynamic stability is directly translated to a rather high temperature for hydrogen release (ca. 300 °C). The enormous stability of saline [MgH2]∞ is mainly due to the rather high lattice energy for the rutile salt structure of [MgH2]∞: the experimentally determined value of ΔH is 2718 kJ mol−1.99 Recent calculations by de Jong et al. have shown that there is a strong influence of cluster size on the thermodynamic stability of (MgH2)n clusters.14 Aggregates with cluster size n < 19 show a sudden drop in stability due to loss in lattice energies (percentually more ions are situated at the surface of the cluster). The calculated temperatures for hydrogen release rapidly decrease with cluster size: for Mg9H18 a decomposition temperature of circa 200 °C has been calculated. Therefore, the question arises whether molecular magnesium hydride complexes could function as model systems for hydrogen storage materials. Although one could envision a possible reversible reaction between [DIPPnacnacMgH]2 and [DIPPnacnacMgI]2 + H2, the dimeric magnesium hydride complex is described as an extremely stable compound (dec. 300 °C) and also the dimeric MgI complex does not react with molecular hydrogen.71 However, Jones et al. found that [DIPPnacnacMgH]2 can be chemically reduced to the MgI dimer with K. The resulting MgI species can be chemically oxidized again to [DIPPnacnacMgH]2 by reaction with AlH3 (cf.eqn (9)).74
Harder et al.78 recently found that the larger magnesium hydride cluster (Fig. 9b) releases molecular hydrogen already at 200 °C (note that this is comparable to the temperature predicted for Mg9H18).14 Gas quantification by Töpler pump measurements quite accurately showed release of all five equivalents of hydrogen. Decomposition experiments with dimeric [DIPPnacnacMgH]2 and a tetranuclear magnesium hydride (Fig. 9a) showed that there is indeed a relationship between cluster size and decomposition temperature. In contrast to earlier findings, the dimer decomposed already at 150 °C, whereas the tetranuclear cluster needed a slightly higher temperature of 175 °C.77 Hitherto, the nature of the decomposition products is unknown, but on account of the reddish colour it is speculated that MgI species could be prevalent. The reversibility of hydrogen storage in molecular magnesium hydride clusters is currently under investigation.
7 Conclusions
Although their synthesis and stabilization is challenging, early main group metal hydride complexes are now an established class of compounds. The arsenal of synthetic methods is steadily growing: whereas PhSiH3 functions as the most frequently used hydride source, molecular H2 or boranes and stannanes also can be used, as can soluble metal hydride compounds like LiAlH4 or AlH3 (and to a certain extent KH). Other methods include the β-hydride elimination route (e.g. for tBu−, iPr2N− or H3B(iPr)NH−). Today, a variety of structures, from monomer, dimer, tetramer to higher aggregates, have been identified by single crystal X-ray diffraction.These metal hydride complexes not only function as reagent of choice in selective reductions but also frequently are proposed to be key intermediates in catalytic cycles in which early main group metal compounds are operative. A thorough understanding of structure, reactivity and stability is therefore immediately of relevance for further development of the field of early main group metal catalysis.
In the light of the growing interest for hydrogen as a future energy source, fundamental understanding of early main group metal hydride complexes should be a valuable asset. Molecular magnesium hydride clusters indeed have been shown to release hydrogen at much lower temperatures than [MgH2]∞ salt itself. Such clusters can be accurately studied at the molecular level and not only exact bonding information but also the first H–Mg–H NMR coupling constants have been observed. It is likely that larger magnesium hydride clusters could function as model systems that facilitate the study of such hydrogen storage systems.
As this new compound class is still relatively new, there is ample room for further investigations on their reactivity, catalytic reactions and other applications. Especially the heavier metal hydrides (Rb, Cs, Sr and Ba), which are most challenging to stabilize, will be the subject of further development in the field. It is anticipated that the novel early main group metal hydride complexes will find increasing interest by the community and that further applications in various fields of chemistry will arise.
Notes and references
- L. Schlapbach and A. Züttel, Nature, 2001, 414, 353 CrossRef CAS.
- G. S. McGrady and G. Guilera, Chem. Soc. Rev., 2003, 32, 383 RSC.
- W. Grochala and P. P. Edwards, Chem. Rev., 2004, 104, 1283 CrossRef CAS.
- S. Aldridge and A. J. Downs, Chem. Rev., 2001, 101, 3305 CrossRef CAS.
- (a) S. Harder, Chem. Rev., 2010, 110, 3852 CrossRef CAS; (b) A. G. M. Barrett, M. R. Crimmin, M. S. Hill and P. A. Procopiou, Proc. R. Soc. A, 2010, 466, 927 CrossRef CAS.
- A. F. Holleman, E. Wiberg and N. Wiberg, Lehrbuch der Anorganischen Chemie, Berlin, 2007, 102nd edn., de Gruyter Search PubMed.
- Handbook of Chemistry and Physics, ed. R. C. Weast and M. J. Astle, CRC Press, Florida, 1981, 61st edn., p. D-94 Search PubMed.
- W. M. Mueller, J. P. Blackledge and G. G. Libowitz, Metal Hydrides, Academic Press, 1968 Search PubMed.
- G. J. Brendel, E. M. Marlett and L. M. Niebylski, Inorg. Chem., 1978, 17, 3589 CrossRef CAS.
- G. S. Smith, Q. C. Johnson, D. K. Smith, D. E. Cox, R. L. Snyder, R.-S. Zhou and A. Zalkin, Solid State Commun., 1988, 67, 491 CrossRef CAS.
- T. Noritake, M. Aoki, S. Towata, Y. Seno and Y. Hirose, Appl. Phys. Lett., 2002, 81, 2008 CrossRef CAS.
- F. A. Cotton and G. Wilkinson, Advanced Inorganic Chemistry, 1988, New York, John Wiley & Sons, p. 119 Search PubMed.
- C. A. Brown, J. Org. Chem., 1974, 39, 3913 CrossRef CAS.
- R. W. P. Wagemans, J. H. van Lenthe, P. E. de Jongh, A. J. van Dillen and K. P. de Jong, J. Am. Chem. Soc., 2005, 127, 16675 CrossRef CAS.
- H. C. Browne and S. Krishnamurthy, Tetrahedron, 1979, 35, 567 CrossRef.
- D. Jacobi, C. Floriani, A. Chiesi-Villa and C. Rizzoli, J. Am. Chem. Soc., 1993, 115, 3595 CrossRef.
- D. Jacobi, C. Floriani, A. Chiesi-Villa and C. Rizzoli, J. Am. Chem. Soc., 1993, 115, 7025 CrossRef.
- N. Etkin, A. J. Hoskin and D. W. Stephan, J. Am. Chem. Soc., 1997, 119, 11420 CrossRef CAS.
- (a) W. W: Lukens Jr, P. T. Matsunaga and R. A. Andersen, Organometallics, 1998, 17, 5240 CrossRef; (b) S. I. Troyanov, V. Varga and K. Mach, Chem. Commun., 1993, 1174 Search PubMed.
- K. D. Conroy, W. E. Piers and M. Parvez, Organometallics, 2009, 28, 6228 CrossRef CAS.
- Z. Hou, Y. Zhang, O. Tardiff and Y. Wakatsuki, J. Am. Chem. Soc., 2001, 123, 9216 CrossRef CAS.
- H. Nöth, A. Schlegel, J. Knizek and H. Schwenk, Angew. Chem., Int. Ed. Engl., 1997, 36, 2640 CrossRef.
- Y. Wang, B. Quillian, P. Wei, C. S. Wannere, P. v. R. Schleyer and G. H. Robinson, Organometallics, 2006, 25, 3286 CrossRef CAS.
- T. Dubé, S. Gambarotta and G. Yap, Organometallics, 2000, 19, 121 CrossRef.
- M. Veith, P. König, A. Rammo and V. Huch, Angew. Chem., Int. Ed., 2005, 44, 5968 CrossRef CAS.
- E. C. Ashby and R. D. Schwartz, Inorg. Chem., 1971, 10, 355 CrossRef CAS.
- P. A. A. Klusener, L. Brandsma, H. D. Verkruijsse, P. v. R. Schleyer, T. Friedl and R. Pi, Angew. Chem., Int. Ed., 1986, 25, 465 CrossRef.
- R. Pi, T. Friedl, P. v. R. Schleyer, P. A. A. Klusener and L. Brandsma, J. Org. Chem., 1987, 52, 4299 CrossRef CAS.
- P. A. A. Klusener, Synthetic Applications of Strongly Basic Reagents, Dissertation, University of Utrecht, 1989 Search PubMed.
- E. R. Burkhardt, C. P. Suddon, J. Brüning and D. F. Rouda, Patent WO, 0071552 (A1) 2000 Search PubMed.
- F. Buch, J. Brettar and S. Harder, Angew. Chem., Int. Ed., 2006, 45, 2741 CrossRef CAS.
- J. L. Brefort, R. Corriu, C. Guérin and B. Henner, J. Organomet. Chem., 1989, 370, 9 CrossRef CAS.
- D. R. Armstrong, W. Clegg, R. P. Davies, S. T. Liddle, D. J. Linton, P. R. Raithby, R. Snaith and A. E. H. Wheatley, Angew. Chem., Int. Ed., 1999, 38, 3367 CrossRef CAS.
- D. R. Armstrong, W. Clegg, R. P. Davies, S. T. Liddle, D. J. Linton, P. R. Raithby, R. Snaith and A. E. H. Wheatley, Angew. Chem., Int. Ed., 1999, 38, 3367 CrossRef CAS.
- S. R. Boss, M. P. Coles, R. Haigh, P. B. Hitchcock, R. Snaith and A. E. H. Wheatley, Angew. Chem., Int. Ed., 2003, 42, 5593 CrossRef CAS.
- S. R. Boss, J. M. Cole, R. Haigh, R. Snaith, A. E. H. Wheatley, G. J. McIntyre and P. R. Raithby, Organometallics, 2004, 23, 4527 CrossRef CAS.
- D. Hoffmann, T. Kottke, R. J. Lagow and R. D. Thomas, Angew. Chem., Int. Ed., 1998, 37, 1537 CrossRef.
- A. Stasch, Angew. Chem., Int. Ed., 2012, 51, 1930 CrossRef CAS.
- U. Blindheim, G. E. Coates and R. C. Srivastava, J. Chem. Soc., Dalton Trans., 1972, 2302 RSC.
- G. E. Coates, D. L. Smith and R. C. Srivastava, J. Chem. Soc., Dalton Trans., 1973, 618 RSC.
- N. A. Bell and G. E. Coates, J. Chem. Soc. A, 1966, 1069 RSC.
- N. A. Bell and G. E. Coates, J. Chem. Soc., 1965, 692 RSC.
- G. E. Coates and M. Tranah, J. Chem. Soc. A, 1967, 236 RSC.
- G. E. Coates and F. Glockling, J. Chem. Soc., 1954, 22, 615 Search PubMed.
- G. W. Adamson, N. A. Bell and H. M. M. Shearer, Acta Crystallogr., 1981, B37, 68 CAS.
- N. A. Bell, G. E. Coates, M. L. Shearer and H. M. M. Shearer, Chem. Commun., 1983, 828 RSC.
- R. Han and G. Parkin, Inorg. Chem., 1992, 31, 983 CrossRef CAS.
- G. E. Coates and J. A. Heslop, J. Chem. Soc. A, 1968, 514 RSC.
- E. C. Ashby and A. B. Goel, Inorg. Chem., 1977, 1441 CrossRef CAS.
- E. C. Ashby and A. B. Goel, J. Org. Chem., 1977, 42, 22 Search PubMed.
- A. B. Goel and E. C. Ashby, J. Organomet. Chem., 1981, 214, C1 CrossRef CAS.
- E. C. Ashby and A. B. Goel, J. Am. Chem. Soc., 1977, 99, 310 CrossRef CAS.
- E. C. Ashby and A. B. Goel, Inorg. Chem., 1978, 17, 1862 CrossRef CAS.
- E. C. Ashby and A. B. Goel, Inorg. Chem., 1979, 18, 1306 CrossRef CAS.
- E. C. Ashby, S. A. Noding and A. B. Goel, J. Org. Chem., 1980, 45, 1028 CrossRef CAS.
- T. P. Hanusa, Coord. Chem. Rev., 2000, 210, 329 CrossRef CAS.
- J. S. Alexander and K. Ruhlandt-Senge, Eur. J. Inorg. Chem., 2002, 11, 2761 CrossRef.
- M. Westerhausen, Coord. Chem. Rev., 2008, 252, 1516 CrossRef CAS.
- (a) F. G. N. Cloke, P. B. Hitchcock, M. F. Lappert, G. A. Lawless and B. Royo, Chem. Commun., 1991, 724 RSC; (b) C. Eaborn, S. A. Hawkes, P. B. Hitchcock and J. D. Smith, Chem. Commun., 1997, 1961 RSC.
- (a) F. Feil and S. Harder, Organometallics, 2000, 19, 5010 CrossRef CAS; (b) S. Harder, F. Feil and A. Weeber, Organometallics, 2001, 20, 1044 CrossRef CAS; (c) S. Harder, F. Feil and K. Knoll, Angew. Chem., Int. Ed., 2001, 40, 4261 CrossRef CAS.
- K. Mochida, Y. Hiraga, H. Takeuchi and H. Ogawa, Organometallics, 1987, 6, 2293 CrossRef CAS.
- J. P. Dunne, M. Tacke, C. Selinka and D. Stalke, Eur. J. Inorg. Chem., 2003, 1416 CrossRef CAS.
- D. J. Gallagher, K. W. Henderson, A. R. Kennedy, C. T. O'Hara, R. E. Mulvey and R. B. Rowlings, Chem. Commun., 2002, 376 RSC.
- P. C. Andrikopoulos, D. R. Armstrong, A. R. Kennedy, R. E. Mulvey, C. T. O'Hara and R. B. Rowlings, Eur. J. Inorg. Chem., 2003, 3354 CrossRef CAS.
- M. J. Harvey, T. P. Hanusa and M. Pink, Chem. Commun., 2000, 489 RSC.
- J. Prust, K. Most, I. Müller, E. Alexopoulos, A. Stasch, I. Úson and H. W. Roesky, Z. Anorg. Allg. Chem., 2001, 627, 2032 CrossRef CAS.
- S. P. Sarish, A. Jana, H. W: Roesky, T. Schulz, M. John and D. Stalke, Inorg. Chem., 2010, 49, 3816 CrossRef.
- S. Harder and J. Brettar, Angew. Chem., 2006, 118, 3554 CrossRef.
- The thallium route, 2 TptBuTl + CaI2 → (TptBu)2Ca gave exclusively the product TptBuCaI: M. H. Chisholm, J. C. Gallucci and K. Phomphrai, Inorg. Chem., 2004, 43, 6717 Search PubMed.
- S. Harder, Organometallics, 2002, 21, 3782 Search PubMed.
- S. P. Green, C. Jones and A. Stasch, Angew. Chem., Int. Ed., 2008, 47, 9079 Search PubMed.
- J. Spielmann, D. F.-J. Piesik and S. Harder, Chem.–Eur. J., 2010, 16, 8307 Search PubMed.
- S. J. Bonyhady, C. Jones, S. Nembenna, A. Stasch, A. J. Edwards and G. J. McIntyre, Chem.–Eur. J., 2010, 16, 938 Search PubMed.
- S. J. Bonyhady, D. Collis, G. Frenking, N. Holzmann, C. Jones and A. Stasch, Nat. Chem., 2010, 2, 865 Search PubMed.
- M. J. Michalczyk, Organometallics, 1992, 11, 2307 Search PubMed.
- M. Arrowsmith, M. S. Hill, D. J. MacDougall and M. F. Mahon, Angew. Chem., Int. Ed., 2009, 48, 4013 Search PubMed.
- S. Harder, J. Intemann and J. Spielmann, mansucript in preparation.
- S. Harder, J. Spielmann, J. Intemann and H. Bandmann, Angew. Chem., Int. Ed., 2011, 50, 4156 Search PubMed.
- M. Arrowsmith, M. S. Hill, G. Kociok-Köhn, D. J. MacDougall and M. F. Mahon, Angew. Chem., Int. Ed., 2012, 51, 2098 Search PubMed.
- P. Jochmann, J. P. Davin, T. P. Spaniol, L. Maron and J. Okuda, Angew. Chem., Int. Ed., 2012, 51, 2098 Search PubMed.
- E. C. Ashby, J. J. Lin and A. B. Goel, J. Org. Chem., 1978, 43, 1564 Search PubMed.
- J. Spielmann and S. Harder, Chem.–Eur. J., 2007, 13, 8928 Search PubMed.
- J. Spielmann and S. Harder, Eur. J. Inorg. Chem., 2008, 1480 Search PubMed.
- J. Spielmann, F. Buch and S. Harder, Angew. Chem., Int. Ed., 2008, 47, 9434 Search PubMed.
- J. Spielmann, G. Jansen, H. Bandmann and S. Harder, Angew. Chem., Int. Ed., 2008, 47, 6290 Search PubMed.
- J. Spielmann and S. Harder, J. Am. Chem. Soc., 2009, 131, 5064 Search PubMed.
- D. J. Liptrot, M. S. Hill, M. F. Mahon and D. J. MacDougall, Chem.–Eur. J., 2010, 16, 8508 Search PubMed.
- J. Spielmann and S. Harder, Dalton Trans., 2011, 40, 8314 Search PubMed.
- P. Bellham, M. S. Hill, D. J. Liptrot, D. J. MacDougall and M. F. Mahon, Chem. Commun., 2011, 47, 9060 Search PubMed.
- C. Jones, S. J. Bonyhady, S. Nembenna and A. Stasch, Eur. J. Inorg. Chem., 2012 Search PubMed early view.
- (a) Z. T. Xiong, C. K. Yong, G. T. Wu, P. Chen, W. Shaw, A. Karkamkar, T. Autrey, M. H. V. K. Diyabalanage, R. P. Shrestha, T. A. Semelsberger, B. L. Scott, M. E. Bowden, B. L. Davis and A. K. Burrell, Angew. Chem., Int. Ed., 2007, 46, 8995 Search PubMed; (b) O. Jones, S. R. Johnson, P. P. Edwards and W. I. F. David, Nat. Mater., 2008, 7, 138 Search PubMed.
- J. Spielmann, M. Bolte and S. Harder, Chem. Commun., 2009, 6934 Search PubMed.
- M. S. Hill, D. J. MacDougall and M. F. Mahon, Dalton Trans., 2010, 39, 11129 Search PubMed.
- (a) K. Ziegler and H. Zeiser, Chem. Ber., 1930, 63, 1847 Search PubMed; (b) D. R. Armstrong, R. E. Mulvey, D. Barr, R. Snaith and D. Reed, J. Organomet. Chem., 1988, 350, 191 Search PubMed; (c) W. Clegg, L. Dunbar, L. Horsburgh and R. E. Mulvey, Angew. Chem., Int. Ed., 1996, 35, 753 Search PubMed.
- M. Arrowsmith, M. S. Hill, T. J. Hadlington, G. Kociok-Köhn and C. Weetman, Organometallics, 2011, 30, 5556 Search PubMed.
- S. Harder and J. Spielmann, J. Organomet. Chem., 2012, 698, 7 Search PubMed.
- M. Arrowsmith, T. J. Hadlington, M. S. Hill and G. Kociok-Köhn, Chem. Commun., 2012, 4567 Search PubMed.
- C. Wu and H.-M. Cheng, J. Mater. Chem., 2010, 20, 5390 Search PubMed.
- C. M. Stander and R. A. Pacey, J. Phys. Chem. Solids, 1978, 39, 829 Search PubMed.
Footnotes |
| † This article is part of the ChemComm ‘Frontiers in Molecular Main Group Chemistry’ web themed issue. |
| ‡ Current address: Anorganische Chemie, Friedrich-Alexander Universitaet Erlangen, Egerlandstrasse 1, 91058 Erlangen, Germany. E-mail: sjoerd.harder@chemie.uni-erlangen.de; Tel: +49 9131 8527350. |
| This journal is © The Royal Society of Chemistry 2012 |