Chiroptical properties of an alternatingly functionalized cellotriose bearing two porphyrin groups†
Keita
Sakakibara
*ab,
Fumiaki
Nakatsubo
c,
Alfred D.
French
d and
Thomas
Rosenau
*a
aDepartment of Chemistry, University of Natural Resources and Life Sciences (BOKU), Muthgasse 18, Vienna A-1190, Austria. E-mail: thomas.rosenau@boku.ac.at; Fax: +43-1-47654 6059; Tel: +43-1-47654 6071
bInstitute for Chemical Research, Kyoto University, Gokasho, Uji, Kyoto 611-0011, Japan. E-mail: sakakibara.keita.4n@kyoto-u.ac.jp; Fax: +81-774-38-3170; Tel: +81-774-38-3168
cResearch Institute for Sustainable Humanosphere, Kyoto University, Gokasho, Uji, Kyoto 611-0011, Japan
dSouthern Regional Research Center, U. S. Department of Agriculture, 1100 Robert E. Lee Blvd, New Orleans, Louisiana 70124, USA
First published on 22nd March 2012
Abstract
Right-handedness derived from bisporphyrins attached to a cellotriose backbone at O-6 and O′′-6 positions is revealed for the first time. This cellotriose is proposed as a model of alternatingly functionalized cellulosics, which have promising properties for applications in optoelectronics and molecular receptors owing to the chirality and rigid backbone effects.
Cellulose, which certainly needs no further introduction as a green and renewable bioresource, combines good physical properties, such as strength and elasticity, and favourable chemical behaviour, such as chemical and thermal stress resistance, in its different material appearances.1 Its derivatives with photoactive properties have become increasingly important.2 At present, regioselective cellulose functionalization and subsequent self-assembling procedures are a reliable approach to produce cellulosic functional nanomaterials. For example, we have reported an efficient artificial photosynthetic system of thin films of 6-O-porphyrinated-2,3-di-O-acylcelluloses and revealed that the attached chromophores as photoelectroactive actuators are spaced at a fixed distance, which is set by the inherent cellulose structure acting as a molecular scaffold, leading to efficient cellulose-based photovoltaics.3 For obtaining advanced cellulosic materials, the following points should be considered and utilized: (1) inherent linearity, rigidity and regularity of the cellulose backbone, inducing equal distances between regioselective substituents, (2) its helicity, possibly inducing supramolecular structures, and (3) the necessity of a precise control of topology and substituent pattern. In their crystal structures cellulose polymorphs are helices with two-fold screw axes, but due to the geometry of the chiral D-glucose building block, cellulose molecules adopt left-handed helical architectures in solution.4 A tendency towards left-handed helices is also observed when geometries from related small molecules are imposed on cellulose chains.5 Thus, chromophores attached to cellulose chains, either covalently or in non-bonded complexes,6 would be helical as well.6 Some of such cellulose derivatives have been applied in practical chiral packing materials for HPLC,7 though details of the formation of chiral helices at all levels are currently far from being sufficiently exploited.
The intriguing potential of alternatingly functionalized celluloses lies in equally spaced functional groups at the repeating distance of 10.3 Å along their axis (Fig. 1a), resulting in promising properties for chiroptical and chiral molecular receptor applications. Reports on such functionalizations are scarce, but Isogai and co-workers have recently reported on the synthesis of an alternating glucose/glucuronic acid co-polysaccharide prepared through TEMPO-oxidation of native celluloses.8
 | ||
| Fig. 1 (a) Alternatingly functionalized cellulose and (b) chemical structure of cellotriose derivative 1. | ||
In this report we communicate the synthesis of an alternatingly functionalized cellotriose (1) with π-electron functionality, porphyrins, which is a suitable model to study the fundamental properties of alternatingly functionalized cellulosics. Compound 1 contains two porphyrins at O-6 and O′′-6 positions, two methyl (Me) groups at the glycosidic end and at O-4′′, and seven benzyl (Bn) groups at the remaining hydroxyl positions (Fig. 1b). For the otherwise deprotected model compounds, the two methyl groups at the proximal reducing end (methyl glycoside) and the terminal 4-O are required to avoid making hydrogen bonds that would not be found in cellulose, by allowing only lateral interactions and suppressing H-bonds from the terminal 4-OH and the glycosidic OH.9 The resulting debenzylated cellotriose, with these two methyl groups representing truncated cellulose chains, thus can be seen as a structural section of a cellulose molecule. Porphyrins were chosen since the exciton-coupled circular dichroic (CD) method10 as well as host–guest complexation with fullerene (C60)11 are well established. Herein, we demonstrate that compound 1 exhibits intramolecular electron coupling as compared to the corresponding monomer 2 (chemical structure is shown in the ESI†) and right-handedness arising from a porphyrin–porphyrin couplet between the two points of attachments, which has not been reported so far. Furthermore, the placement of the substituents on the first and third residues provides an optimum receptor function for C60, inducing an electronic interaction in the π-space environment between the two chromophores.
The synthetic details of the path towards compound 1 are given in the ESI.† As depicted in Scheme 1, the cellotriose structure was produced by a glycosidation coupling of a novel cellobiosyl donor activated by a trichloroacetimidoyl group at the C-1 position (donor; 3) and a known glucose-based receptor with a free 4-OH group (acceptor; 6) under Schmidt conditions (summarized in Table S1, ESI†). The reaction proceeded in a satisfying way, yielding 78% of β-configured cellotriose 4, when 0.2 eq. of BF3/Et2O at −30 °C were employed. The key intermediate 5 was prepared through the liberation of primary hydroxyl groups at C-6 and C-6′′ by CAN oxidation in a 44% yield. The coupling of 5 with porphyrin carboxylic acid (TPP–COOH) gave the desired compound 1 in a 71% yield.
 | ||
| Scheme 1 Synthesis of cellotriose derivative 1. | ||
Target 1 was comprehensively analytically characterized. The 1H and 13C NMR spectra, recorded in CDCl3, are shown in Fig. S1, ESI.† The doublets at 4.29, 4.54, and 4.67 ppm of the 1H NMR spectrum with their 3JH,H coupling constants of approx. 8 Hz are assigned to the characteristic protons at C-1, C-1′, and C-1′′, respectively, indicative of β-configuration throughout. Another characteristic of the 1H NMR is that three Bn aromatic protons are shifted upfield (Δδ = ca. 0.5 ppm), arising from extensive aromatic ring-current shielding. This suggests that pronounced π–π stacking interactions between the two porphyrin moieties are formed even though the porphyrins can rotate freely in solution, presumably due to the straightness and stiffness of the bulkily substituted (benzylated) cellotriose chain. The carbon signals corresponding to two porphyrin carbonyls and to the carbons of porphyrinated (C-6 and 6′′) and benzylated (C-6′) glucose units prove the alternating substitution pattern.
Fig. 2a shows the UV–VIS absorption spectra of 1 and 2 in CHCl3. Compound 1 has a typical intense Soret band (λmax = 419 nm) and five satellite Q bands (between 490 and 700 nm) were evident, which are due to π–π* transitions of the conjugated macrocycles. The Soret band shows a reduced molar absorption coefficient slightly blue-shifted by ca. 2 nm as compared to the corresponding monomer 2 (λmax = 421 nm) owing to the interaction of neighboring porphyrin molecules. This is also observed in the Q-band regions. It should be noted that the spectrum of 1 shows a new shoulder band at 450 nm. A similar observation was reported by Qiu and co-workers12 and Redl and co-workers.4f They described that a J-type aggregate structure leads to such a shoulder band. On the other hand, the CHCl3 solution of 1 was diluted (2.5 μM) and appeared clear without any turbid substance. It seems appropriate, therefore, that this band at 450 nm would be due to the intramolecular electron coupling between the two porphyrin moieties, which is likely the result of the conformational restriction because of the nature of the rigid oligosaccharide backbone.
 | ||
| Fig. 2 (a) UV–VIS and (b) CD spectra of 1 in CHCl3 (blue) and that of the corresponding glucose derivative 2 in CHCl3 (red); c = 2.5 μM (UV–VIS) and 5.0 μM (CD). Δε is quoted per chromophoric unit. (c) Proposed right-handed helicity of the bisporphyrins on the basis of the CD spectrum. | ||
The CD spectrum for 1 dissolved in CHCl3 (5 μM) is shown in Fig. 2b. In spite of the fact that the porphyrin chromophores are not attached directly to the chiral carbons of the anhydroglucopyranose units, a positive bisignate CD curve with a maximum molar circular dichroism (Δε) of 12.3 L mol−1 cm−1 per chromophoric unit at 427 nm was observed, corresponding to the right-handed helical arrangement of the chromophores. This exciton splitting observed in the CD spectrum could be explained by some extent of the conformational rigidity as well as enough distance (10.3 Å) between adjacent porphyrins of 1, since the porphyrin moieties are not free to assume many orientations even in solution. It should be emphasized that the positive cotton effect (Fig. 2c) has never been observed so far in the reports of cellulose derivatives that give rise to CD bands exhibiting exciton splitting. For example, Harkness and Gray reported that the CD spectrum of 6-O-α-(1-naphtylmethyl)-2,3-di-O-pentylcellulose in cyclo-hexane exhibits a weak negative CD band at 228 nm.4b Similarly, Redl and co-workers reported the electron coupling between the porphyrin moieties attached to the methylcellulose chains at the C-6 position, observing the negative Cotton effect with a maximum Δε of ca. 200 L mol−1 cm−1 per chromophoric unit at about 420 nm.4f Since this value is much larger than the Δε of 1, it can be safely concluded that the exciton-coupled CD of alternatingly functionalized celluloses has not been recognized so far because of the weak intensity, and the right-handedness derived from alternatingly substituted porphyrins on the cellotriose backbone is revealed for the first time.
Analogous to cellulose, the backbone of the cellotriose segment would take a left-handed twist, which was confirmed by QM minimizations of 1. The values of the interglycosidic torsion angles ϕ and ψ were within the observed range for crystal structures.5 At the same time, the porphyrin groups, as demonstrated herein, have a right-handed helical character, which puts the question how a left-handed cellulose helix can give rise to a right-handed chromophore helix? The reason for the opposite handedness is the placement of the porphyrins on every second anhydroglucopyranose unit. Consider a 3-fold cellulose helix with perfect 3-fold screw-axis symmetry (Fig. 3): a left-handed helix is generated when the glucose residues are each rotated −120° and advanced 5 Å along the helix axis from their predecessor's positions. The porphyrin residues are rotated −240° since they are on every second residue only. At the same time, by the rules of helix definition, one would define this helix by rotating instead +120°, giving a right-handed helix of chromophores (Fig. 3). These theoretical considerations explain and confirm the above experimental observations.
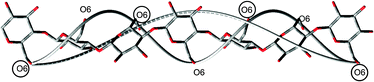 | ||
| Fig. 3 Schematic drawing of a cellulose helix that is left-handed, with exactly three anhydroglucopyronose (AHG) residues per turn of the helix. Two turns are shown, so that the first, fourth and seventh AHG residue all have the same orientation. Each O6 is labeled, and every other O6 has a circle around it to represent a porphyrin substituent. Also shown are two “helical threads” that connect the O6 atoms.5 (A similar thread could connect any particular type of atom, as long as the molecule is a helix.) One of the threads connects all of the O6 atoms, and it is left-handed. The other helix only connects the circled O6 atoms, and it is right-handed. Since the CD measurements detect only the chromophoric porphyrin rings at every second AHG unit, a right-handed helix is reported, even though the helical thread through all of the O6 (in every AHG unit) is left-handed. | ||
To demonstrate the receptor ability of 1, supramolecular complexes formed between the porphyrin tweezers host and fullerene (C60) were analyzed. Fig. 4a presents the UV–Vis spectra of 1 in CHCl3 in the presence of incremental amounts of C60, exhibiting a substantial decrease of the Soret band as well as an increase of a band at 450 nm. Since the van der Waals diameter of C60 is about 1 nm,13 π–π stacking (sandwich formation) between the bisporphyrins in 1 and fullerene is likely (Fig. 4b). While these results are preliminary, they do show that alternatingly functionalized celluloses are expected to become a novel type of synthetic molecular receptors owing to their inherent scaffolding properties.
 | ||
| Fig. 4 (a) UV–VIS absorption changes of 1 (c = 2.5 μM) upon addition of C60 (1, 2, 3, 4, and 5 eq.) in CHCl3. The absorbance of fullerene had been subtracted from the spectra. (b) Schematic representation of the C60 assembly with 1. | ||
In conclusion, we have demonstrated for the first time the right-handedness of an alternatingly porphyrinated cellotriose. This finding provides an excellent argument in favor of the high application potential of alternatingly functionalized celluloses, and a good strategy for the preparation of the corresponding model compounds.
The authors thank Dr C. Obinger and Dr P. Furtmueller (BOKU) for support for CD measurement and Dr A. Hofinger (BOKU) for support for NMR measurement. The financial support by the Austrian BMLFUW Ministry (Project “Multifunctional celluloses”) is gratefully acknowledged. K.S. acknowledges the Foundation of Yoshida Science and Technologies.
Notes and references
- (a) D. Klemm, B. Heublein, H.-P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358 CrossRef CAS; (b) Y. Nishio, Adv. Polym. Sci., 2006, 205, 97 CrossRef CAS.
- H. Wondraczek, A. Kotiaho, P. Fardim and T. Heinze, Carbohydr. Polym., 2011, 83, 1048 CrossRef CAS.
- (a) K. Sakakibara, Y. Ogawa and F. Nakatsubo, Macromol. Rapid Commun., 2007, 28, 1270 CrossRef CAS; (b) K. Sakakibara and F. Nakatsubo, Macromol. Chem. Phys., 2008, 209, 1274 CrossRef CAS; (c) K. Sakakibara and F. Nakatsubo, Macromol. Chem. Phys., 2010, 211, 2429 CrossRef.
- (a) B. R. Harkness and D. G. Gray, Macromolecules, 1990, 23, 1452 CrossRef CAS; (b) B. R. Harkness and D. G. Gray, Macromolecules, 1991, 24, 1800 CrossRef CAS; (c) Y. Nishio, Y. Tani, N. Kimura, H. Suzuki, S. Ito, M. Yamamoto, B. R. Harkness and D. G. Gray, Macromolecules, 1995, 28, 3818 CrossRef CAS; (d) E. Yashima, J. Noguchi and Y. Okamoto, Macromolecules, 1995, 28, 8368 CrossRef CAS; (e) R. Aburatani, Y. Okamoto and K. Hatada, Bull. Chem. Soc. Jpn., 1990, 63, 3606 CrossRef CAS; (f) F. X. Redl, M. Lutz and J. Daub, Chem.–Eur. J., 2001, 7, 5350 CrossRef CAS.
- A. D. French and G. P. Johnson, Cellulose, 2004, 11, 5 CrossRef CAS.
- (a) A. R. Engle, N. Purdie and J. A. Hyatt, Carbohydr. Res., 1994, 265, 181 CrossRef CAS; (b) A. M. Ritcey and D. G. Gray, Biopolymers, 1988, 27, 479 CrossRef CAS.
- Y. Okamoto, J. Polym. Sci., Part A-1: Polym. Chem., 2008, 47, 1731 Search PubMed.
- M. Hirota, K. Furihata, T. Saito, T. Kawada and A. Isogai, Angew. Chem., Int. Ed., 2010, 49, 7670 CrossRef CAS.
- (a) I. D. Mackie, J. Röhrling, R. O. Could, J. Pauli, C. Jäger, M. Walkinshaw, A. Potthast, T. Rosenau and P. Kosma, Carbohydr. Res., 2002, 337, 161 CrossRef CAS; (b) M. del Carmen Ruiz Ruiz, J. Querner, I. Adorjan, P. Kosma and T. Rosenau, Macromol. Symp., 2006, 232, 68 CrossRef.
- (a) S. Matile, N. Berova, K. Nakanishi, S. Novkova, I. Philipova and B. Blagoev, J. Am. Chem. Soc., 1995, 117, 7021 CrossRef CAS; (b) X. Li and B. Borhan, J. Am. Chem. Soc., 2008, 130, 16126 CrossRef CAS.
- (a) K. Tashiro, T. Aida, J.-Y. Zheng, K. Kinbara, K. Saigo, S. Sakamoto and K. Yamaguchi, J. Am. Chem. Soc., 1999, 121, 9477 CrossRef CAS; (b) Z.-Q. Wu, X.-B. Shao, C. Li, J.-L. Hou, K. Wang, X.-K. Jiang and Z.-T. Li, J. Am. Chem. Soc., 2005, 127, 17460 CrossRef CAS; (c) N. Berova, G. Pescitelli, A. G. Petrovic and G. Proni, Chem. Commun., 2009, 5958 RSC.
- Y. Qiu, P. Chen and M. Liu, J. Am. Chem. Soc., 2010, 132, 9644 CrossRef CAS.
- W. Krätschmer, L. D. Lamb, K. Fostiropoulos and D. R. Huffman, Nature, 1990, 347, 354 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental procedures, synthesis, NMR spectra, supporting figures and table. See DOI: 10.1039/c2cc30805c |
| This journal is © The Royal Society of Chemistry 2012 |