Blending Baeyer–Villiger monooxygenases: using a robust BVMO as a scaffold for creating chimeric enzymes with novel catalytic properties†
Hugo L.
van Beek
,
Gonzalo de
Gonzalo
and
Marco W.
Fraaije
*
Laboratory of Biochemistry, Groningen Biomolecular Sciences and Biotechnology Institute, University of Groningen, Nijenborgh 4, 9747 AG, Groningen, The Netherlands. E-mail: m.w.fraaije@rug.nl; Tel: +31 50-363-4345
First published on 10th January 2012
Abstract
The thermostable Baeyer–Villiger monooxygenase (BVMO) phenylacetone monooxygenase (PAMO) is used as a scaffold to introduce novel selectivities from other BVMOs or the metagenome by structure-inspired subdomain exchanges. This yields biocatalysts with new preferences in the oxidation of sulfides and the Baeyer–Villiger oxidation of ketones, all while maintaining most of the original thermostability.
Baeyer–Villiger monooxygenases (BVMOs) represent a unique class of oxidative enzymes that are able to perform Baeyer–Villiger oxidations and sulfoxidations, often with high chemo-, regio-, and/or enantioselectivity.1 In the last few years a number of novel BVMOs have been identified and studied for their use as biocatalysts. However, only one of the BVMOs employed was shown to be robust: phenylacetone monooxygenase (PAMO).2 PAMO is relatively thermostable, displays activity at a broad pH range, and tolerates various organic solvents and ionic liquids.3 Unfortunately, PAMO shows a narrow substrate specificity, only accepting small aromatic ketones, sulfides, amines and boron compounds.4 This is in contrast with the most thoroughly studied BVMO, cyclohexanone monooxygenase (CHMO) from Acinetobacter sp., for which hundreds of substrates have been reported.1,5
Both CHMO and PAMO have been the subject of enzyme engineering studies to alter their substrate specificity and enantioselectivity.6 Crystal structures of PAMO and CHMO are available to guide these enzyme engineering efforts.7 These have revealed that mutations in a loop at the substrate binding site (residues 440–446) have profound effects on substrate specificity and enantioselectivity.6a,8 So far, engineering attempts on PAMO have focused on the conversion of novel substrates by introducing a limited number of mutations close to the active site.
To expand the substrate range of PAMO, we set out to blend the substrate specificities of sequence-related BVMOs into PAMO. Steroid monooxygenase (STMO) and CHMO display a high degree of sequence similarity (53% and 39% sequence identity) with PAMO,9 while they accept a much wider substrate range. STMO converts small aromatic compounds like phenylacetone and thioanisole 1 but also the relatively bulky substrate such as progesterone 5. CHMO has been reported to act on a wide range of aliphatic compounds. As the sequence identities are too low for a classical gene shuffling approach,10 we decided to blend enzyme properties by performing structure-inspired subdomain exchanges. For this, the C-terminal part of PAMO was replaced by the respective subdomains of other BVMOs, including the 440–446 loop and part of the second sphere residues (Fig. 1). By blending BVMOs in this way, a large part of the thermostable PAMO is conserved, while a significant part of the substrate binding site is exchanged with that of another BVMO. It was anticipated that PAMO, due to its inherent stability, could act as a stable scaffold to accommodate a subdomain of another BVMO.
 | ||
| Fig. 1 Schematic view of the design of BVMO blends. PAMO is shown in green, the complementary BVMO subdomains in blue, magenta and yellow, respectively. | ||
Subdomains were exchanged by introducing a suitable restriction site in the PAMO gene, allowing ligation-independent cloning.11 The single targeted crossover ensured an in-frame recombination while combining two genes with limited sequence identity. A limited set of chimeric BVMOs was created, allowing us to focus on the expansion of substrate specificity, instead of screening a large combinatorial library for conversions of a limited number of compounds. Specifically, the 106 C-terminal residues of PAMO were exchanged with the homologous regions of STMO or CHMO to create PASTMO and PACHMO, respectively. The other created chimera, PAMEMO1, contained the C-terminal part of a putative BVMO (53% sequence identity with PAMO) for which the gene has been sequenced as part of a metagenome sequencing effort.12 The metagenome sequence database has so far hardly been exploited for novel biocatalysts and with this study we wanted to explore the potential of this source for BVMO activities. All three BVMO chimeras were fused to a thermostable and His-tagged phosphite dehydrogenase (PTDH) at the N-terminus.13 This facilitated effective expression and purification of the created BVMO blends while the fused PTDH can be exploited for cofactor regeneration.14 All created chimeric BVMOs were successfully overexpressed as soluble proteins. Purification revealed that all proteins contained the flavin cofactor, indicative of proper protein folding. To probe the stability of all created BVMO-blends, the ThermoFAD method was employed.15 Melting temperatures of the chimeric BVMOs were lower (49–55 °C) when compared to PAMO (61 °C), but significantly higher than those of STMO and CHMO (both 39 °C) (Table 1). This shows that the subdomain exchange yields stable BVMO variants. The inverse chimeras, CHPAMO and STPAMO, were also created. However, these chimeric proteins could not be expressed, probably due to the relatively labile N-terminal scaffold.
| Entry | Biocatalyst | T M (°C) | Conv.b,c (%) | eeb (%) | Config. |
|---|---|---|---|---|---|
| a 5.0 mM substrate incubated with 10 μM BVMO at pH 7.5 and 24 °C. Reaction time 24 h. b Determined by GC. c Only small amounts of the sulfone overoxidation product (≤5%) were observed. | |||||
| 1 | PAMO | 61 | 93 | 16 | R |
| 2 | STMO | 39 | 87 | 27 | S |
| 3 | CHMO | 39 | 70 | ≥99 | R |
| 4 | PASTMO | 49 | 79 | 62 | S |
| 5 | PACHMO | 55 | 45 | 60 | R |
| 6 | PAMEMO1 | 51 | 82 | 60 | S |
Different compounds were used to compare the activity and specificity of the PAMO-based chimeric BVMOs with the parent enzymes (Scheme 1). The substrates were chosen in such a way that effects on substrate specificity, regio- and/or enantioselectivity could be assessed. All chimeric BVMOs prepared were shown to be active on most of the tested compounds (see Scheme 1).
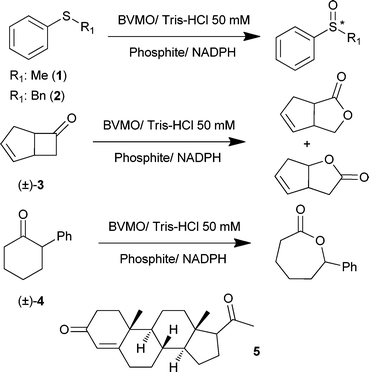 | ||
| Scheme 1 Substrates tested in the biocatalyzed oxidations using different wild-type and chimeric Baeyer–Villiger monooxygenases. | ||
Thioanisole 1 was used as a prochiral substrate to probe the enantioselectivity of native and chimeric BVMOs (Table 1). PAMO and CHMO preferentially form the (R)-sulfoxides with poor and excellent enantioselectivity, respectively (entries 1 and 2). The enantioselectivity of the PAMO–CHMO hybrid behaves as a blend of the parent enzymes as it forms the (R)-sulfoxide with moderate enantioselectivity, as shown in entry 5. STMO is the only native BVMO which is able to form the (S)-enantiomer, albeit with moderate enantioselectivity. Interestingly, PASTMO and PAMEMO1 exhibited improved S enantioselectivities respecting wild type STMO, showing that the exchanged subdomain plays a dominant role in the enzymatic selectivity.
The bulkier compound benzyl phenyl sulfide 2 was also tested to probe the effect of BVMO blending. While this sulfide was not accepted by both STMO and CHMO, incubation with PAMO led to poor conversion and selectivity (7% (R)-sulfoxide with 36% enantiomeric excess (ee) after 48 h). Chimera PASTMO did not convert this substrate. Conversion by PACHMO resulted in 14% of predominantly the (R)-enantiomer with a good enantioselectivity (70% ee). Intriguingly, oxidation of 2 by PAMEMO1 resulted in the most effective conversion (39% sulfoxide formed), generating the opposite S enantiomer with a moderate optical purity (56% ee).
In view of the promising results in the sulfoxidation processes, we analyzed different racemic ketones. Bicyclic ketone (±)-bicyclo[3.2.0]hept-2-en-6-one (±)-3 has often been used to investigate the stereoselectivity of BVMOs because four distinct chiral products can be formed from this compound. The so-called normal lactones are formed when the oxygen is inserted next to the tertiary carbon, as opposed to the secondary carbon for the abnormal lactones. This gives two chemically distinct products for both the (1S,5R)- and (1R,5S)-enantiomers. This conversion provides information about differences in regio- and enantioselectivities of the different biocatalysts. The three chimeric BVMOs were found to be just as effective as the native BVMOs in converting (±)-3 (Table 2, entries 1–3). PASTMO shows an intermediate enantioselectivity when compared with the parent enzymes, while attaining the regioselectivity of STMO. Interestingly, PACHMO displays a small preference for the abnormal lactone, something that is not observed with any of the parent BVMOs. Both major enantiomers are obtained with complete enantioselectivity. The introduction of the metagenomic gene fragment renders a PAMO variant, PAMEMO1, which shows novel properties by forming predominantly the normal lactone with excellent selectivity (entry 6). These data show that by the design of chimeric BVMOs, thermostable PAMO variants can be made that display novel catalytic properties when compared with native BVMOs.
| Entry | Biocatalyst | Conv.b (%) | Ratio norm./abn. | ee (1S,5R) normalb (%) | ee (1R,5S) abnormalb (%) |
|---|---|---|---|---|---|
| a 5.0 mM substrate incubated with 10 μM BVMO at pH 7.5 and 24 °C. Reaction time 24 h. b Determined by GC. | |||||
| 1 | PAMO | 60 | 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 :50 |
29 | ≥99 |
| 2 | STMO | 58 | 65:35 |
79 | 7 |
| 3 | CHMO | 69 | 50:50 |
97 | ≥99 |
| 4 | PASTMO | 75 | 66:34 |
76 | 62 |
| 5 | PACHMO | 38 | 39:61 |
≥99 | ≥99 |
| 6 | PAMEMO1 | 58 | 76:24 |
≥99 | 49 |
For a further exploration of the created BVMO blends, also the racemic ketone (±)-2-phenylcyclohexanone (±)-4 was tested (Table 3). The corresponding enantiomerically pure lactones formed represent attractive building blocks in organic chemistry. It has been shown before that CHMO is capable to form the (R)-lactone with excellent optical purity (entry 1). In contrast, we found that STMO does not act on this ketone while PAMO forms the (S)-enantiomer with poor selectivity (entry 3). The use of chimeric enzymes PASTMO and PAMEMO1 led to the formation of the (S)-lactones with good (PASTMO) to excellent enantioselectivities, as observed in entries 5 and 6 for PACHMO and PAMEMO1. Conversions achieved with the chimeric enzymes are equal to or higher than that obtained with wild type PAMO.
| Entry | Biocatalyst | Conv.b (%) | ee lactoneb (%) | Config. |
|---|---|---|---|---|
| a 10.0 mM substrate incubated with 4 μM BVMO at pH 7.5 and 24 °C. Reaction time 24 h. b Determined by GC. c Reaction performed with a substrate concentration of 5.0 mM and 10 μM BVMO. | ||||
| 1 | PAMO | 11 | 22 | S |
| 2 | STMO | ≤3 | — | — |
| 3 | CHMO | 42 | ≥99 | R |
| 4 | PASTMO | 30 | 70 | S |
| 5 | PACHMO | 11 | ≥99 | R |
| 6 | PAMEMO1c | 24 | ≥99 | R |
Finally, progesterone 5 was tested as substrate of different BVMOs. This compound was oxidized by STMO with a high conversion (90% after 24 hours), while neither PAMO nor CHMO was capable of oxidizing this steroid. Incubation of 5 with PACHMO and PAMEMO1 led to the recovery of the starting material, while PASTMO afforded the final lactone with a moderate conversion (19% after 24 hours), indicating that this hybrid enzyme can improve the PAMO results for this substrate while retaining its thermostability.
The data above show that exchange of the C-terminal subdomain of PAMO is an effective approach to generate BVMOs that are relatively robust while exhibiting novel catalytic properties. This method of generating novel BVMOs by blending a robust BVMO with gene fragments of other BVMOs is different from the commonly employed methodologies of enzyme engineering or discovery. PAMO appears to be a very suitable scaffold for such a subdomain exchange approach as it allows the exchange of sequences with only 39% sequence identity while retaining thermostability, even after replacing more than 10% of its residues. All purified chimeras were more thermostable than the parent BVMO from which the C-terminal subdomain originated. The method also allows blending of gene fragments from genes that can be selected by an in silico search of (meta)genome sequence databases, exploring untapped sequence space. This permits a very rapid and relatively cheap access to new BVMO activities as such short gene fragments can be easily prepared by modern DNA synthesis protocols.
The substrate specificity of the BVMO blends was found to be at least partially based on the introduced C-terminal part. All chimeras show novel catalytic behaviour, especially concerning enantioselectivity. Not all activities from the parent BVMOs could be introduced in the chimeras. For example, conversion of cyclohexanone was not observed for PACHMO. On the other hand some of the chimeras show unanticipated behaviour by converting substrates with a higher yield or enantiomeric excess than either of both parent enzymes. Thus, PASTMO shows a better conversion and enantioselectivity with the relatively bulky ketones (±)-3 and (±)-4 when compared to the parent enzymes. The results obtained show that the catalytic properties of chimeras are not just the average of those of the parents, but that new properties can be introduced, not present in either of the parents. This represents a starting point to expand the possibilities for BVMO-catalyzed biocatalysis, by creating a portfolio of BVMO blends with suitable stability and expression, and with varying substrate specificities and regio- and/or enantioselectivities.
This work was supported by the EU-FP7 Oxygreen project.
Notes and references
- Some recent reviews: (a) H. Leisch, K. Morley and P. C. K. Lau, Chem. Rev., 2011, 111, 4165 CrossRef CAS; (b) G. de Gonzalo, M. D. Mihovilovic and M. W. Fraaije, ChemBioChem, 2010, 11, 2208 CrossRef CAS; (c) H. Dudek, D. E. Torres Pazmiño and M. W. Fraaije, Curr. Opin. Chem. Biol., 2010, 14, 138 CrossRef CAS.
- M. W. Fraaije, J. Wu, D. P. H. M. Heuts, E. W. van Hellemond, J. H. Lutje Spelberg and D. B. Janssen, Appl. Microbiol. Biotechnol., 2005, 66, 393 CrossRef CAS.
- (a) F. Secundo, S. Fialá, M. W. Fraaije, G. de Gonzalo, M. Meli, F. Zambianchi and G. Ottolina, Biotechnol. Bioeng., 2011, 108, 491 Search PubMed; (b) C. Rodríguez, G. de Gonzalo, M. W. Fraaije and V. Gotor, Green Chem., 2010, 12, 2255 RSC; (c) G. de Gonzalo, G. Ottolina, F. Zambianchi, M. W. Fraaije and G. Carrea, J. Mol. Catal. B: Enzym., 2006, 39, 91 CrossRef CAS.
- P. B. Brondani, G. de Gonzalo, M. W. Fraaije and L. H. Andrade, Adv. Synth. Catal., 2011, 353, 2169 Search PubMed.
- M. D. Mihovilovic, B. Müller and P. Stanetty, Eur. J. Org. Chem., 2002, 22, 3711 CrossRef.
- For example: (a) H. M. Dudek, G. de Gonzalo, D. E. Torres Pazmiño, P. Stepniak, L. S. Wyrwicz, L. Rychlewski and M. W. Fraaije, Appl. Environ. Microbiol., 2011, 77, 687 Search PubMed; (b) S. Wu, J. P. Acevedo and M. T. Reetz, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 2775 CrossRef CAS; (c) M. D. Mihovilovic, F. Rudroff, A. Winninger, T. Schneider, F. Schluz and M. T. Reetz, Org. Lett., 2006, 8, 1221 CrossRef CAS.
- (a) R. Orru, H. M. Dudek, C. Martinoli, D. E. Torres Pazmiño, A. Royant, M. Weik, M. W. Fraaije and A. Mattevi, J. Biol. Chem., 2011, 286, 29284 Search PubMed; (b) I. A. Mirza, B. J. Yachnin, S. Wang, S. Grosse, H. Bergeron, A. Imura, H. Iwaki, Y. Hasegawa, P. C. Lau and A. M. Berghuis, J. Am. Chem. Soc., 2009, 131, 8848 Search PubMed; (c) E. Malito, A. Alfieri, M. W. Fraaije and A. Mattevi, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 13157 CrossRef CAS.
- (a) M. T. Reetz and S. Wu, J. Am. Chem. Soc., 2009, 131, 15424 CrossRef CAS; (b) M. T. Reetz and S. Wu, Chem. Commun., 2008, 5499 RSC; (c) D. E. Torres Pazmiño, R. Snajdrova, D. V. Rial, M. D. Mihovilovic and M. W. Fraaije, Adv. Synth. Catal., 2007, 349, 1361 CrossRef; (d) M. Bocola, F. Schulz, F. Leca, A. Vogel, M. W. Fraaije and M. Reetz, Adv. Synth. Catal., 2005, 347, 979 CrossRef CAS.
- M. Miyamoto, J. Matsumoto, T. Iwaya and E. Itagaki, Biochim. Biophys. Acta, 1995, 1251, 115 Search PubMed.
- W. P. C. Stemmer, Nature, 1994, 370, 389 CrossRef CAS.
- B. Zhu, G. Cai, E. O. Hall and G. J. Freeman, BioTechniques, 2007, 43, 354 Search PubMed.
- J. Gilbert, Annu. Rev. Mater. Sci., 2011, 3, 347 Search PubMed.
- D. E. Torres Pazmiño, R. Snajdrova, B. J. Braas, M. Ghrobial, M. D. Mihovilovic and M. W. Fraaije, Angew. Chem., Int. Ed., 2008, 47, 2275 CrossRef.
- J. M. Vrtis, A. K. White, W. W. Metcalf and W. A. van der Donk, J. Am. Chem. Soc., 2001, 123, 2672 CrossRef CAS.
- F. Forneris, R. Orru, D. Bonivento, L. R. Chiarelli and A. Mattevi, FEBS J., 2009, 276, 2833 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: All experimental procedures are included in the supporting information. See DOI: 10.1039/c2cc17656d |
| This journal is © The Royal Society of Chemistry 2012 |