Simplifying native chemical ligation with an N-acylsulfonamide linker†
Fabienne
Burlina
a,
Caroline
Morris
b,
Raymond
Behrendt
c,
Peter
White
*d and
John
Offer
*b
aUPMC Univ Paris 06, CNRS, ENS, UMR 7203 Laboratoire des Biomolécules, 4 place Jussieu, 75005 Paris, France
bDivision of Physical Biochemistry, MRC National Institute for Medical Research, Mill Hill, London, NW7 1AA, UK. E-mail: joffer@nimr.mrc.ac.uk; Tel: +44 2088162082
cMerck & Cie, Im Laternenacker 5, 8200 Schaffhausen, Switzerland
dMerck Chemicals, Padge Road, Beeston, Notts, NG9 2JR UK. E-mail: peter.white@merckgroup.com
First published on 31st January 2012
Abstract
We report a simplified procedure for the chemical ligation of peptides by using the sulfamylbutyryl linker as a mildly activating group capable of participating in ligation. When the peptidyl N-methylsulfonamide is directly added with excess thiols to ligation reactions, the speed of reaction is comparable to native chemical ligation.
The use of native chemical ligation (NCL) has revolutionized the synthesis and semi-synthesis of small proteins.1 NCL is the coupling of two unprotected peptides, one having an N-terminal cysteine and the other a C-terminal thioester to give an amide bond at the ligation site. The mildly activated peptide thioesters are generally synthesized by the Boc/benzyl method using in situ neutralization protocols.2,3 However, the requirement for anhydrous HF and the increased difficulties in obtaining HF have driven chemists to look for alternatives.4,5 Furthermore, demand for phosphorylated or glycosylated peptide thioesters or other acid-sensitive modifications that preclude the use of HF has led to increased activity in this search.6 The problem of developing a robust Fmoc thioester method is part of the broader problem of synthesising a C-terminal active ester peptide using Fmoc SPPS. The deprotection of the Fmoc group with base at each cycle is not compatible with an active ester at the C-terminus. Many ingenious approaches have been developed to generate the required thioester peptide.4,5,7 However, the most popular has been to use an N-acylsulfonamide as a base and acid stable (safety-catch) linker for peptide synthesis. Alkylation of the sulfonamide after peptide assembly makes the linker labile to cleavage with nucleophiles.8 The first application of this approach to peptide thioester preparation was described by Pessi and co-workers,9 wherein, following activation, the fully-protected peptide was cleaved from the resin by sodium thiophenolate in DMF. The resulting protected peptide thioester was subsequently treated with acid to remove side-chain protection. The use of the sulfonamide linker has allowed the synthesis of several impressive targets, including long peptide thioesters10glycoproteins11 and phosphoproteins.6,12,13 However, the thiolysis step can sometimes be problematic because of poor solvation of the resin bound peptide and difficulties with peptide recovery from DMF.
We wanted to explore the ligation properties of N-peptidyl- N-alkylsulfonamides because, although poorly reactive with nucleophiles14 we considered that for both stability during purification and chemoselectivity a weakly activating group was desirable. The reactivity could theoretically be modulated by the addition of thiols to the ligation milieu. The possibility of unprotected N-peptidyl-N-alkylsulfonamides participating directly in ligation would expand the applicability of NCL and remove the thioester conversion steps (Scheme 1). Unprotected N-peptidyl-N-alkylsulfonamides are easy to access by Fmoc SPPS by scalable, robust procedures and they are very stable compounds.
 | ||
| Scheme 1 Peptide ligation with an N-acylsulfonamide component. Pep 1 and 2 correspond to unprotected peptides. | ||
To prepare peptideN-alkylsulfonamides the sulfamylbutyryl linker was attached to an acid-labile resin, in this case Sieber amide resin. Acid cleavage after alkylation would give the N-alkylsulfonamide derivatised peptide (Scheme 2). An additional advantage, previously reported, is that it allows the monitoring of the alkylation state of the sulfonamide by LC-MS on a small portion of resin.15 In our case, however, the principle consideration for using a double linker strategy was to derivatise peptides with the sulfonamide so it acts as a mildly activated functional group.
 | ||
| Scheme 2 Preparation of the peptidyl-N-methylsulfonamide. | ||
A complication with the use of the sulfamylbutyryl linker is incomplete acylation on solid phase of the poorly nucleophilic sulfonamide by the first amino acid. This has been overcome here by derivatisation of the linker with the C-terminal residue prior to its attachment to the solid phase (ESI, Scheme S1).
Another key consideration for this approach was the choice of alkylating agent. The alkylation reaction has to be quantitative to prevent complicating HPLC purification after cleavage. In addition, the alkyl group has an effect on the reactivity of the sulfonamide,14 the less reactive the sulfonamide, the smaller the risk of self-condensation during purification and of unwanted side reactions during ligation. Therefore methylation was selected. Trimethylsilyl diazomethane (TMS-CHN2) after Pessi and co-workers9,16,17 was used for methylation with the solvent mix hexane/DCM (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) identified by Mezzato et al.15
:1) identified by Mezzato et al.15
To evaluate our approach, we prepared model N-peptidylN-methylsulfonamides 1 (H-AYRAG- N(CH3)SO2(CH2)3CONH2) and 2 (H-AYRAA- N(CH3)SO2(CH2)3CONH2) to explore their ligation properties with an N-terminal cysteine peptide. Fmoc-Gly-sulfamylbutyric acid and Fmoc-Ala-sulfamylbutyric acid were synthesised (ESI, Scheme S1) and coupled to the Sieber Amide resin. Peptide chain extension was carried out with standard Fmoc SPPS protocols, except the N-terminal residue, was incorporated as a Boc-protected amino acid. Alkylation, typically performed overnight with TMS-CHN2 was checked by cleaving a small sample of each resin and analysing the peptides by HPLC and MALDI-TOF mass spectrometry (ESI). Where the reaction was incomplete it was continued and monitored with further sample cleavage until completion.15 Standard cleavage of the bulk resins gave peptides 1 and 2.
Ligation in the most favoured case between peptide 1 (glycine terminal) and H-CFALRGWR-NH2 (3) at high concentration (10 mM each) was conducted in neutral aqueous buffer and in the presence of 4-mercaptophenylacetic acid (MPAA). Gratifyingly, ligation was found to occur rapidly (approx. t1/2 = 30 min). The efficacy of our approach was further tested using peptide 2 that contains the more sterically challenging C-terminal residue, alanine. The reaction was complete in 2 h (Fig. 1), indicating that N-peptidyl-N-methylsulfonamides are effective substitutes for pre-formed thioesters in NCL reactions.
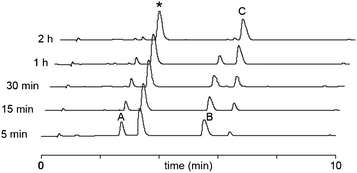 | ||
| Fig. 1 Analytical HPLC time course of the ligation reaction of AYRAA N-methylsulfonamide to CFALRGWR. Ligation conditions: peptide (10 mM each), 200 mM sodium phosphate buffer pH 7.5, 2 mM EDTA, 1 M Guanidine.HCl, 50 mM TCEP, 60 mM MPAA, 40 °C. HPLC conditions: Chromolith column, 0–30% B (0.1% TFA, 90% CH3CN) in 10 min, 3 ml.min−1. A = H-AYRAA-N(CH3)SO2(CH2)3CONH2, B = H-CFALRGWR-NH2 C = ligation product H-AYRAACFALRGWR-NH2, * MPAA. | ||
The ligation mechanism probably occurs via the thioester formed by exchange of the N-peptidyl-N-methylsulfonamide with MPAA. This thioester was observed in the absence of N-teminal cysteine peptides but not in their presence, giving very clean HPLC profiles for the ligation reaction (Fig. 1 and 2). This is in contrast to standard NCL with preformed alkyl thioesters, where the monitoring of the reaction by HPLC is often complicated by the presence of multiple thioester exchange products. In the sulfonamide ligation, the MPAA thioester formed is instantly captured by cysteine, a better nucleophile than MPAA. A similar observation was made by Melnyk and co-workers, who performed ligation reactions at high MPAA concentration with mildly activated peptides.7
![BPTI ligation (a) MALDI-TOF mass spectrum of the ligation product. (b) Analytical HPLC time course of the ligation. Ligation conditions: [BPTI(1–37)N(CH3)SO2(CH2)3CONH2] = 0.65 mM, [BPTI(38–58)] = 1 mM in 200 mM phosphate buffer pH 7.5, 2 mM EDTA, 6 M Guanidine.HCl, 50 mM TCEP, 60 mM MPAA, 40 °C. HPLC conditions: RP-C18, 0–70% B in 25 min, 1 ml.min−1. A = BPTI(1–37)N(CH3)SO2(CH2)3CONH2, B = BPTI(38–58), C = ligation product, * MPAA.](/image/article/2012/CC/c2cc15911b/c2cc15911b-f2.gif) | ||
| Fig. 2 BPTI ligation (a) MALDI-TOF mass spectrum of the ligation product. (b) Analytical HPLC time course of the ligation. Ligation conditions: [BPTI(1–37)N(CH3)SO2(CH2)3CONH2] = 0.65 mM, [BPTI(38–58)] = 1 mM in 200 mM phosphate buffer pH 7.5, 2 mM EDTA, 6 M Guanidine.HCl, 50 mM TCEP, 60 mM MPAA, 40 °C. HPLC conditions: RP-C18, 0–70% B in 25 min, 1 ml.min−1. A = BPTI(1–37)N(CH3)SO2(CH2)3CONH2, B = BPTI(38–58), C = ligation product, * MPAA. | ||
Small, model peptides are often imperfect guides for how larger peptides ligate because of their low molecular weight and limited functionality. Model ligations are typically performed at high concentrations of approximately 10 mM; however most syntheses of small proteins are carried out at around 1 mM because of the higher molecular weights of the peptides used. Therefore we undertook the synthesis of a classic target for chemical synthesis, BPTI.18,19 This 58 residue, small protein is a stringent test of the acylsulfonamide approach with its full range of side chain functional groups. The length is more typical of peptides used for NCL. We adopted the strategy of Lu and co-workers and prepared BPTI by the ligation of two fragments: BPTI(1–37) and BPTI (38–58).19 BPTI(38–58) was assembled on Fmoc-Ala-Wang resin using standard HOBt/DIC activation. BPTI(1–37) was assembled in a similar manner on Fmoc-Gly-sulfamylbutyryl Sieber amide resin (ESI). The linker was methylated with TMS-CHN216 monitoring with HPLC till completion and cleaved with TFA to afford deprotected BPTI(1–37)-N-methylsulfonamide. Following purification by RP-HPLC, the two fragments were ligated under denaturing conditions (Fig. 2) using low millimolar fragment concentration. The reaction was almost complete in 8 h and was comparable to the classical NCL using preformed thioester of Lu and co-workers.19
In conclusion, we have demonstrated a variation on the sulfamylbutyryl linker approach that overcomes many of the current limitations, and provides a simpler method for peptide ligation viaFmoc SPPS. The sulfamylbutyryl group acts as a latent thioester. The notoriously poor reactivity of N-peptidyl-N-methylsulfonamides is in this context advantageous as it minimises unwanted side reactions during purification and ligation and additionally its stability is promising for long term storage.14 This development, of a proven methodology that is compatible with the multiple functionalities and conditions used in peptide synthesis should prove to be valuable for the synthesis of the chemically defined post-translationally modified proteins currently demanded by biological research.
This work was supported by a grant-in-aid from the MRC number U117592730.
Notes and references
- S. B. Kent, Chem. Soc. Rev., 2009, 38, 338–351 RSC
.
- T. M. Hackeng, J. H. Griffin and P. E. Dawson, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 10068–10073 CrossRef CAS
- M. Schnolzer, P. Alewood, A. Jones, D. Alewood and S. B. Kent, Int. J. Pept. Protein Res., 1992, 40, 180–193 CrossRef CAS
- J. Kang and D. Macmillan, Org. Biomol. Chem., 2010, 8, 1993–2002 CAS
- F. Mende and O. Seitz, Angew. Chem., Int. Ed., 2011, 50, 1232–1240 CrossRef CAS
- J. Li, I. A. Taylor, J. Lloyd, J. A. Clapperton, S. Howell, D. MacMillan and S. J. Smerdon, J. Biol. Chem., 2008, 283, 36019–36030 CrossRef CAS
- N. Ollivier, J. Dheur, R. Mhidia, A. Blanpain and O. Melnyk, Org. Lett., 2010, 12, 5238–5241 CrossRef CAS
- G. W. Kenner, J. R. McDermott and R. C. Sheppard, J. Chem. Soc., Chem. Commun., 1971, 636–637 RSC
- R. Ingenito, E. Bianchi, D. Fattori and A. Pessi, J. Am. Chem. Soc., 1999, 121, 11369–11374 CrossRef CAS
- F. Mende, M. Beisswenger and O. Seitz, J. Am. Chem. Soc., 2010, 132, 11110–11118 CrossRef CAS
- D. Macmillan and C. R. Bertozzi, Angew. Chem., Int. Ed., 2004, 43, 1355–1359 CrossRef CAS
- M. Huse, M. N. Holford, J. Kuriyan and T. W. Muir, J. Am. Chem. Soc., 2000, 122, 8337–8338 CrossRef CAS
- R. R. Flavell, M. Huse, M. Goger, M. Trester-Zedlitz, J. Kuriyan and T. W. Muir, Org. Lett., 2002, 4, 165–168 CrossRef CAS
- B. J. Backes, A. A. Virgilio and J. A. Ellman, J. Am. Chem. Soc., 1996, 118, 3055–3056 CrossRef CAS
- S. Mezzato, M. Schaffrath and C. Unverzagt, Angew. Chem., Int. Ed., 2005, 44, 1650–1654 CrossRef CAS
- Caution, trimethylsilyl diazomethane is extremely toxic. N. G. Murphy, S. M. Varney, J. M. Tallon, J. R. Thompson and P. D. Blanc, Clin. Toxicol., 2009, 47, 712 Search PubMed
- This reagent does, however, methylate phosphate diesters, such as the monobenzyl esters employed for the introduction of phosphoamino acids during Fmoc-based SPPS. Fortunately, this side reaction can be reversed by using TMSBr/TFA for the cleavage of the peptide from the solid phase. E. A. Kitas, J. W. Perich, R. B. Johns and G. W. Tregear, Tetrahedron Lett., 1988, 29, 3591–3592 CrossRef CAS
- M. Ferrer, C. Woodward and G. Barany, Int. J. Pept. Protein Res., 1992, 40, 194–207 CrossRef CAS
- W. Lu, M. A. Starovasnik and S. B. Kent, FEBS Lett., 1998, 429, 31–35 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c2cc15911b |
| This journal is © The Royal Society of Chemistry 2012 |