Decarboxylative benzylation and arylation of nitriles†‡
Antonio
Recio, III
,
Jeffrey D.
Heinzman
and
Jon A.
Tunge
*
2010 Malott Hall, 1251 Wescoe Hall Dr, Lawrence, KS USA. E-mail: tunge@ku.edu; Fax: 785-864-5396; Tel: 785-864-4136
First published on 7th November 2011
Abstract
Decarboxylative benzylation of nitriles is achieved via coupling of metallated nitriles with Pd-π-benzyl complexes that are generated in situ from cyanoacetic benzyl esters. In addition, decarboxylative couplings of α,α-disubstituted 2-methylfuranyl cyanoacetates can lead to either decarboxylative arylation or benzylation depending on the reaction conditions.
Recently there has been much interest in developing catalytic decarboxylative cross-coupling reactions as alternatives to traditional cross-coupling reactions.1 In 2009, our group reported that the decarboxylative allylation of cyanoacetic esters (DMSO pKa ∼ 22–33)2 proceeded by in situ generation of a metallated nitrile species under formally neutral conditions.3,4 Traditionally, generation of metallated nitriles takes place under highly basic conditions, requiring a metal hydride, alkyl lithiate,5 or lithium amide base.6,7 Alternatively, Flemming, Knochel and others8 have accessed metallated nitrilesvia treatment of α-halo nitriles with Grignard reagents.9 The resulting metallated nitriles are readily alkylated with a variety of electrophiles, including benzyl halides.6
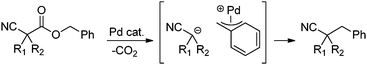
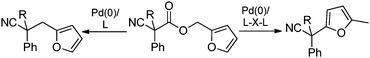
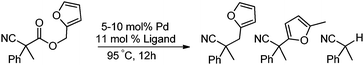
In our efforts to develop methods for benzylation that occur under mild conditions and avoid the use to toxic benzylic halides,10 we hypothesized that decarboxylative benzylation11,12 would allow one to synthesize benzylated nitriles from activated benzyl alcohol derivatives. Herein, we report the decarboxylative benzylation of nitrilesvia the likely intermediacy of Pd-π-benzyl complexes (eqn (1)).12 In addition, we disclose that appropriate modification of the palladium catalyst allows one to achieve either decarboxylative benzylation or decarboxylative arylation of nitriles with furans derived from furylmethyl esters (eqn (2)).
 | (1) |
 | (2) |
Our initial studies began by determining competent catalytic conditions for achieving decarboxylative benzylation (DcB) of nitriles with “simple” aromatic and heteroaromatic benzyl esters. Previous reports involving the DcB of alkynes and ketones suggested that Pd-π-benzyl formation was difficult with benzyl esters that lacked extended conjugation, so coupling of simple benzene-derived esters was difficult.11a Interestingly, our initial studies revealed that treatment of a benzyl cyanoacetate with Pd(PPh3)4 resulted in oxidative addition of the benzyl ester, but decarboxylation was followed by protonation to form NCCH(Me)Ph rather than the desired C–C bond formation (Table 1, entry 1). Gratifyingly, conditions developed by Hiyama for cross-coupling of benzyl carbonates (cond. B)12a proved to be excellent for formation of Pd-π-benzyl complexes, allowing for efficient carbon–carbon bond formation with simple benzyl alcohol derivatives (e.g.Table 1, entry 2).

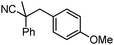
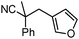
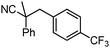

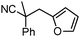
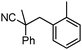
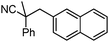
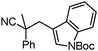
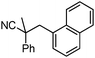
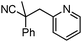
To briefly probe the scope of benzyl electrophiles that are compatible with decarboxylative benzylation, a variety of α-methyl-α-phenyl cyanoacetates were synthesized and subjected to the standard reaction conditions. Interestingly, substrates with electron-donating substituents (Table 1, entry 3) and electron-withdrawing functionalities (entries 4, 5) both provided benzylated products in good yields. The DcB reaction was not affected by ortho-substitution (entry 6) providing excellent conversion to the benzylated product. Furthermore, both the α-methyl- and β-methyl naphthyl cyanoesters (entries 7, 8) were competent substrates for DcB coupling.
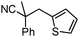
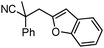
Given the pharmaceutical relevance of heterocyclic arenes,13 we investigated arylmethylations utilizing several heteroaromatic benzyl alcohol moieties. Indeed, treatment of α,α-disubstituted 2-methyl thiophenyl, 3-methyl thiophenyl and 3-methyl furanyl benzyl esters (Table 1, entries 9–11) resulted in smooth conversion to the arylmethylated products in good yields. Other heteroaromatic benzyl cyanoesters were subjected to the reaction conditions listed in Table 1: as shown, 2-methyl benzofuran (entry 13), N-boc protected 3-methyl indole (entry 14), and 2-methyl pyridine (entry 15) were all converted to the arylmethylated products, albeit with dramatically reduced yields.
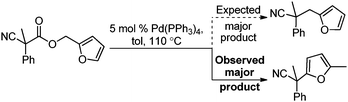 | (3) |
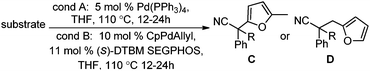
Surprisingly, subjecting the α,α-disubstituted 2-methyl furanyl cyanoacetic esters to similar reaction conditions resulted in formation of a mixture of arylmethylation and arylation products, with the latter being the major product (eqn (3)). This result was quite interesting given that there are no reports for the inter-molecular decarboxylative arylation of nitriles or any other nucleophiles viaPd-π-benzyl complexes, nor is there any precedent for the α-arylation of non-stabilized nitriles under formally neutral conditions.14 Earlier this year, Kwong reported the decarboxylative arylation of nitriles, however did so via the more conventional base mediated coupling of aryl halides.15,16
Intrigued by this result, we turned our attention to developing conditions for both the decarboxylative arylmethylation and arylation of the α,α-disubstituted 2-methyl furanyl cyanoesters. While Pd(PPh3)4 catalyst favored the arylation products (Table 2, entry 1), combining CpPd(allyl) precatalyst with (diphenylphosphino)ethane heavily favored formation of the protonation product (entry 2). Unfortunately, the conditions reported by Hiyama for Pd-π-benzyl couplings also formed significant amounts of the unwanted protonation product (entry 3).12a Having previously shown that BINAP ligand can partially circumvent protonation3 in allylations of sulfones,17 the (rac)-BINAP modified catalyst was investigated and shown to greatly increase the amount of the observed benzylated products.11b Lastly, use of the bulky bidentate phosphine (S)-DTBM SEGPHOS favored formation of the arylmethylation products, with minimal protonation (entry 5, Table 2). Since the screening was conducted with enantioenriched (S)-DTBM SEGPHOS, the product was analyzed by chiral stationary phase HPLC. Unfortunately, the arylmethylated product was formed in low ee (11%).
| Entry | Pd source | Ligand | Solvent |
benzyl.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :aryl.:prot. :aryl.:prot. |
|---|---|---|---|---|
| a Chiral stationary phase chromatography: 11% ee. | ||||
| 1 | Pd(PPh3)4 | None | THF | 14:85:1 |
| 2 | CpPd(allyl) | dppe | THF | <5:<5:>90 |
| 3 | CpPd(allyl) | dppf | THF | 45:<10:45 |
| 4 | CpPd(allyl) | BINAP | THF | 85:<5:10 |
| 5 | CpPd(allyl) | (S) DTBMSEGPHOS | THF |
89
a
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :11:trace :11:trace
|
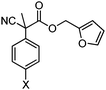
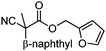
With the newly found reaction conditions, Pd(PPh3)4 to facilitate arylation and CpPd(allyl)/(S)-DTBM SEGPHOS for generation of the arylmethylated products, we then investigated a number of α,α-disubstituted 2-methyl furanyl cyanoester substrates for selectivity (Table 3). Our forays began with examining the electronics of the nitrile anion via synthesis of benzyl esters derivatized at the para-position on the α-phenyl substituent. The results shown in Table 3 suggest that there is no obvious correlation of selectivity with the electronics of the nitrile (entries 1–7). Similarly, a substrate with a β-naphthyl substituent provides the arylated product with Pd(PPh3)4, and arylmethylation product when using the bidentate SEGPHOS derivative. In addition, only small changes in selectivities were observed when exchanging the α-methyl group for more sterically demanding α-benzyl or isopropyl substituents (Table 3, entries 10–16). Lastly, substituents with functional groups that are capable of coordinating to the catalyst (alkene, pyridine) reduced the selectivity for arylation (entries 14, 17). Moreover, these substrates did not undergo C–C bond formation under conditions that were expected to afford arylmethylated product (cond. B).
| Entry | Substrate | Conditions | C:Da |
% Yieldb | |
|---|---|---|---|---|---|
| a Calculated from crude HNMR. b Combined yield (isolated yield major isomer). c Contains 5% protonation byproduct. d Contains 8% protonation byproduct. | |||||
| 1 |
|
X = H | A | 84:14 |
86 (71) |
| 2 | B | 11:89 |
83 (65) | ||
| 3 | X = OMe | A | >95:<5 |
75 (75) | |
| 4 | B | <5:>95 |
69 (69)c | ||
| 5 | X = Cl | A | 80:20 |
84 (69) | |
| 6 | B | 10:90 |
— (65) | ||
| 7 | X = CN | A | >95:<5 |
70 (70) | |
| 8 |
|
A | >95:<5 |
89 (89) | |
| 9 | B | 16:84 |
70 (53) | ||
| 10 |
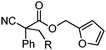
|
R = Ph | A | 90:10 |
— (75) |
| 11 | B | 9:91 |
86 (77)c | ||
| 12 | R = 4-OMePh | A | 90:10 |
84 (70) | |
| 13 | B | <5:>95 |
76 (76)d | ||
| 14 | R = 2-Cl-5-pyridyl | A | 75:25 |
65 (51) | |
| 15 |
|
R= | A | 85:15 |
90 (63) |
| 16 | i-propyl | B | <5:>95 |
71 (71) | |
| 17 | R = Allyl | A | 75:25 |
— (60) |
In simplest terms, the results in Table 3 represent a distinct switch in selectivity when changing from the monodentate PPh3 ligand to the bidentate DTBM-SEGPHOS ligand. We postulate that both products originate from formation of an η3-Pd-π-furfuryl intermediate.18 To explain the ligand-dependant selectivity, we suggest that monodentate ligand (PPh3) allows access to an open coordination site on the metal center, allowing for inner-sphere attack of the nucleophile as suggested in Scheme 1, I. Hartwig has crystallographically characterized an analogous palladium ketiminate complex,16c and mechanistic studies of an allylative dearomatization reaction by Lin19 suggest that our proposed η3-π-benzyl, η1-N-bound ketenimine3,20 transition state is feasible.21,22 Moreover, dearomatization of benzyl electrophiles with allenyl stannanes likely proceeds by intermediates similar to I.21b Lack of an available coordination site with the bidentate ligated system forces outer-sphere attack of the nitrile-stabilized anion (Scheme 2, II), delivering the arylmethylated product 8.
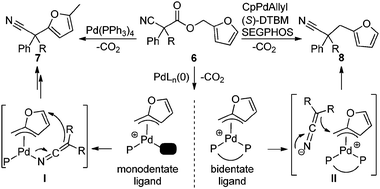 | ||
| Scheme 1 Mechanistic rationale for arylation v. benzylation. | ||
In conclusion, we have developed catalytic decarboxylative benzylations and arylations of nitriles. These methods allow for the coupling of aromatic- and heteroaromatic benzyl alcohol derivatives opposed to traditional alkylations that utilize benzyl halides.
This work has been supported financially the National Institute of General Medical Sciences (NIGMS 1R01GM079644).
Notes and references
-
(a) J. D. Weaver, A. Recio III, A. J. Grenning and J. A. Tunge, Chem. Rev., 2011, 111, 1846 CrossRef CAS
; (b) L. J. Gooßen, F. Collet and K. Gooßen, Isr. J. Chem., 2010, 50, 617 CrossRef
-
(a) F. G. Bordwell, P. Van Der Puy and N. R. Vanier, J. Org. Chem., 1976, 41, 1885 CrossRef CAS
- A. Recio, III and J. A. Tunge, Org. Lett., 2009, 11, 5630 CrossRef
-
(a) J. Tsuji, T. Yamada, I. Minami, M. Yuhara, M. Nisar and I. Shimizu, J. Org. Chem., 1987, 52, 2988 CrossRef CAS
- F. F. Fleming and B. C. Shook, Tetrahedron, 2002, 58, 1 CrossRef CAS
- S. Arseniyadis, K. S. Kyler and D. S. Watt, Org React. (Hoboken, NJ, US), 1984, 31, 1 CAS
- T. Takeda, K. Ando, A. Mamada and T. Fujiwara, Chem. Lett., 1985, 1149 CrossRef CAS
- N. A. Strotman, S. Sommer and G. C. Fu, Angew. Chem., Int. Ed., 2007, 46, 3556 CrossRef CAS
- Knochel and Fleming:
(a) F. F. Fleming, Z. Zhang, W. Liu and P. J. Knochel, J. Org. Chem., 2005, 70, 2200 CrossRef CAS
- S. Marchini, L. Passerini, M. D. Hoglund, A. Pinno and M. Nendza, Environ. Toxicol. Chem., 1999, 18, 2759 CAS
-
(a) R. R. P. Torregrosa, Y. Ariyarathna, K. Chattopadhyay and J. A. Tunge, J. Am. Chem. Soc., 2010, 132, 9280 CrossRef CAS
-
(a) Y. Nakao, S. Ebata, J. Chen, H. Imanka and T. Hiyama, Chem. Lett., 2007, 36, 606 CrossRef CAS
-
(a) A. D. Mills, M. Z. Nazer, M. Haddadin and M. Kurth, J. Org. Chem., 2006, 71, 2687 CrossRef CAS
- R. Shintani, T. Tsuji, S. Park and T. Hayashi, Chem. Commun., 2010, 46, 1697 RSC
- P. Y. Yeung, K. H. Chung and F. Y. Kwong, Org. Lett., 2011, 13, 2912 CrossRef CAS
-
(a) S. Duez, S. Bernhardt, J. Heppekausen, F. F. Fleming and P. Knochel, Org. Lett., 2011, 13, 1690 CrossRef CAS
- J. D. Weaver and J. A. Tunge, Org. Lett., 2008, 10, 4657 CrossRef CAS
- R. D. Dewhurst, R. Müller, M. Kaupp, K. Radacki and K. Götz, Organometallics, 2010, 29, 4431 CrossRef CAS
- A. Ariafard and Z. Lin, J. Am. Chem. Soc., 2006, 128, 13010 CrossRef CAS
-
(a) J. A. Keith, D. C. Behenna, J. T. Mohr, S. Ma, S. C. Marinescu, J. Oxgaard, B. M. Stoltz and W. A. Goddard III, J. Am. Chem. Soc., 129, 11876 CrossRef CAS
-
(a) M. Bao, H. Nakamura and Y. Yamamoto, J. Am. Chem. Soc., 2001, 123, 759 CrossRef CAS
- Nucleophilic dearomatization naphthalene derivatives: B. Peng, S. Zhang, X. Yu, X. Feng and M. Bao, Org. Lett., 2011, 13, 5402 CrossRef CAS
Footnotes |
| † This article is part of the ChemComm ‘Advances in catalytic C–C bond formation via late transition metals’ web themed issue. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c1cc16011g |
| This journal is © The Royal Society of Chemistry 2012 |