Synthesis of 3-substituted 1,5-aldehyde estersvia an organocatalytic highly enantioselective conjugate addition of new carbonylmethyl 2-pyridinylsulfone to enals†‡
Jing
Deng
a,
Fei
Wang
b,
Wenzhong
Yan
a,
Jin
Zhu
a,
Hualiang
Jiang
a,
Wei
Wang
*ac and
Jian
Li
*a
aSchool of Pharmacy, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, China. E-mail: jianli@ecust.edu.cn; Fax: +86 21-64252584; Tel: +86 21-64252584
bChinese Pharmacopoeia commission, Building 11, Fahua Nanli, Beijing, 100061, China
cDepartment of Chemistry and Chemical Biology, University of New Mexico, MSC03 2060, Albuquerque, NM 87131-0001, USA. E-mail: wwang@unm.edu; Fax: +1 505-277-2609
First published on 7th November 2011
Abstract
A highly enantioselective organocatalytic protocol for conjugate addition of new nucleophilic carbonylmethyl 2-pyridinylsulfone to enals has been developed in good yields and with high enantioselectivities. The resulting Michael adducts are versatile building blocks for a variety of organic transformations.
Despite the fact that chiral 3-substituted 1,5-aldehyde esters are versatile building blocks in organic synthesis, for example, they can be conveniently transformed into synthetically and biologically interesting lactones and lactams,1 their synthesis remains elusive.2Conjugate additions to enals represent a straightforward approach to the substances and in the past several years impressive organocatalytic approaches have been reported.3 It is noteworthy that in general in these processes, activated methylenes such as malonates,4 1,3-ketoesters,5nitroalkanes,6 bis-sulfones,7 and active thioesters8 as nucleophiles are commonly used.9 However, the introduction of a more synthetically useful mono-ester (e.g.CH2CO2Me) moiety is an unmet synthetic issue.
We envision that introducing a temporary “mask” group such as a sulfone moiety10–12 instead of a carbonyl can function as a nucleophile, which can be viewed as a CH2CO2Me synthon (Scheme 1). The introduction of the sulfone group kills two birds with one stone. It activates the α-carbon of ester allowing for an effective nucleophilic conjugate addition. Moreover, the experience of ours9 and several others10–12 in this area shows that the moiety can be readily removed under mild reaction conditions without affecting enantioselectivity of the end products. Toward this end, a series of carbonylmethyl sulfones 113 including new carbonylmethyl 2-pyridinylsulfone 1a (CMPS) have been developed and explored for the purpose. We have found that the new reagent 1a is the most effective nucleophile in the organocatalytic Michael addition reaction with enals. Notably, high enantioselectivities (89–>99% ee) are achieved. Moreover, the Michel adducts can be conveniently transformed into new functional molecules and the sulfone group can be removed under mild reactions without affecting enantioselectivity of the products. Herein we disclose the results.
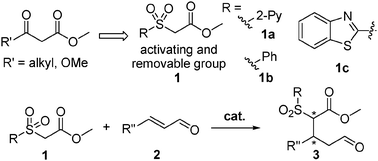 | ||
| Scheme 1 Design of carbonylmethyl sulfones 1 as nucleophiles for organocatalytic Michael reactions of enals. | ||
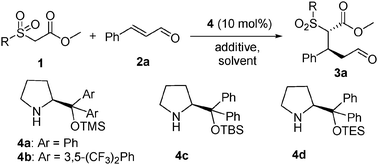
In our initial efforts, three different methoxycarbonylmethyl heterocyclic sulfones 1a–c, which bear 2-pyridinyl, phenyl and benzothiazoyl sulfone groups, were screened in the reaction with trans-cinnamaldehyde 2a in the presence of diphenylprolinol silyl ether 4a (10 mol%),14 a commonly used promoter in conjugate addition to enals in THF at rt (Table 1, entries 1–3). We were pleased to find that the novel methoxycarbonylmethyl 2-pyridinylsulfone 1a showed the highest activity among them tested. Even without a base, the reaction proceeded smoothly to provide the desired product 3a in 72% yield and with 95% ee and 8.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr after 48 h (Table 1, entry 1). Screening of solvents revealed that the yield, enantio- and diastereoselectivities of the products varied dramatically (entries 1, 4–6). Among the solvents probed, toluene was of choice for the process (entry 4). In this instance, a high yield (86%), excellent ee (>99%) and good dr (14.3:1) were achieved. It was noteworthy that methylene dichloride gave the better dr (20:1), but at the expense of the enantioselectivity (91% ee) and yield (53%) (entry 5). When methanol was employed, the yield (90%) was improved but the enantioselectivity (77%) decreased significantly (entry 6). The effect of additives on reactions was also examined (entries 7–9). Among the acids, bases and salts probed, it appeared that they were not beneficial. PhCO2H and LiCl resulted in decreased dr (entries 7 and 8), while Et3N reduced the yield (entry 9). Survey of catalysts revealed that the more bulky catalysts 4b–d could promote this reaction by further improving the dr value to an excellent level (dr > 30:1) (entries 10–12). Among them, catalyst 4d was the best for the process (Table 1, entry 12). In this case, a high yield (90%), high ee (>99%) and excellent dr (>30:1) were obtained. Reducing the loading of 2a from 2.0 equiv. to 1.2 equiv. gave a similar result (entry 14).
:1 dr after 48 h (Table 1, entry 1). Screening of solvents revealed that the yield, enantio- and diastereoselectivities of the products varied dramatically (entries 1, 4–6). Among the solvents probed, toluene was of choice for the process (entry 4). In this instance, a high yield (86%), excellent ee (>99%) and good dr (14.3:1) were achieved. It was noteworthy that methylene dichloride gave the better dr (20:1), but at the expense of the enantioselectivity (91% ee) and yield (53%) (entry 5). When methanol was employed, the yield (90%) was improved but the enantioselectivity (77%) decreased significantly (entry 6). The effect of additives on reactions was also examined (entries 7–9). Among the acids, bases and salts probed, it appeared that they were not beneficial. PhCO2H and LiCl resulted in decreased dr (entries 7 and 8), while Et3N reduced the yield (entry 9). Survey of catalysts revealed that the more bulky catalysts 4b–d could promote this reaction by further improving the dr value to an excellent level (dr > 30:1) (entries 10–12). Among them, catalyst 4d was the best for the process (Table 1, entry 12). In this case, a high yield (90%), high ee (>99%) and excellent dr (>30:1) were obtained. Reducing the loading of 2a from 2.0 equiv. to 1.2 equiv. gave a similar result (entry 14).
| Entry | 1 | Cat. | Solvent | Additive | Yieldb (%) | drc | eed (%) |
|---|---|---|---|---|---|---|---|
| a Unless stated otherwise, the reaction was carried out with 1 (0.2 mmol), 2a (0.4 mmol), catalyst 4 (0.02 mmol) and additive (0.02 mmol) in 1 mL of solvent at rt for 48 h. b Isolated yield. c The diastereoisomeric ratios were determined by 1H NMR of products. d Determined by HPLC analysis (Chiralcel AD) by converting to the corresponding methylacetal. e Reaction time: 72 h. f 2a (0.3 mmol) was used. g 2a (0.24 mmol) was used. | |||||||
| 1 | 1a | 4a | THF | None | 72 | 8.3:1 |
95 |
| 2 | 1b | 4a | THF | None | 54 | 2.3:1 |
91 |
| 3e | 1c | 4a | THF | None | 0 | — | — |
| 4 | 1a | 4a | Toluene | None | 86 | 14.3:1 |
100 |
| 5 | 1a | 4a | CH2Cl2 | None | 53 | 20:1 |
91 |
| 6 | 1a | 4a | MeOH | None | 90 | 1.7:1 |
77 |
| 7 | 1a | 4a | Toluene | PhCO2H | 87 | 6.7:1 |
>99 |
| 8 | 1a | 4a | Toluene | LiCl | 91 | 2.5:1 |
100 |
| 9 | 1a | 4a | Toluene | Et3N | 80 | 14.3:1 |
100 |
| 10 | 1a | 4b | Toluene | None | 40 | >30:1 |
100 |
| 11 | 1a | 4c | Toluene | None | 88 | >30:1 |
100 |
| 12 | 1a | 4d | Toluene | None | 90 | >30:1 |
100 |
| 13f | 1a | 4d | Toluene | None | 90 | >30:1 |
100 |
| 14g | 1a | 4d | Toluene | None | 90 | >30:1 |
100 |
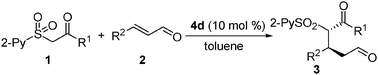
Having established optimal reaction conditions, we subsequently explored the scope of the Michael addition of CMPS 1 to structurally diverse α,β-unsaturated aldehydes. As shown in Table 2, a number of trans-cinnamaldehydes bearing electron-donating and electron-withdrawing substituents were successfully applied in the Michael reaction. The corresponding Michael adducts 3a–o were isolated with high to excellent enantioselectivities (89–>99% ee) and in good to excellent yields (54–95%) (entries 1–18). It seems that independent of the electronic characteristics of the substituents attached to the phenyl ring are the enantioselectivities of the processes. Also significant is that heteroaromatic enals could effectively engage in the conjugate addition process with high efficiency (entry 3) when the reaction was performed at 0 °C. Furthermore, the 4d-promoted Michael process was expanded to the less reactive alkyl α,β-unsaturated aldehyde (entries 4–7). Excellent enantioselectivities (90–94% ee) were obtained despite relative low yields (50–65%) with 20 mol% 4d and K2CO3 as additive. The low reaction yields are mainly due to an significant side 1,2-addition-dehydration reaction between 1a and 2.15 In addition to sulfone ester (1a), the ketones arylcarbonylmethyl 2-pyridinylsulfone (1d) and alkylcarbonylmethyl 2-pyridinylsulfone (1e) also efficiently participate in the conjugate process to generate 1,5-ketoaldehydes (entries 19 and 20). Again, high yields (76 and 94%) and excellent diastereo- and enantioselectivity (>30:1 dr and >99% ee) were achieved. It should be pointed out that although poor diastereoselectivities were seen in some cases, the removal of the sulfone group would eliminate a stereogenic center. We found that (see below) the high enantioselectivity arises from the β-carbon, which is directly controlled by the 4d-mediated conjugate addition. The absolute configuration of the Michael adducts was determined and confirmed by a single crystal X-ray crystallographic analysis of the derivative 3j (Fig. S1, ESI‡).16
| Entry | R1 | R2 | 3 | Yieldb (%) | drc | eed (%) |
|---|---|---|---|---|---|---|
| a Unless stated otherwise, see ESI. b Isolated yield. c Determined by 1H NMR of products. d Determined by HPLC analysis (Chiralcel AD and AS) by converting to the corresponding methylacetal. e At 0 °C. f 10% K2CO3 and 20% catalyst 4d used. g Reaction time: 72 h. | ||||||
| 1 | OMe | Ph | 3a | 90 | >30:1 |
100 |
| 2 | OMe | 4-MeOC6H4 | 3b | 83 | 3:1 |
100 |
| 3e | OMe | 2-Furyl | 3c | 55 | 1.7:1 |
89 |
| 4f,g | OMe | C3H7 | 3d | 54 | 1.8:1 |
92 |
| 5f | OMe | C4H9 | 3e | 65 | 1.8:1 |
94 |
| 6f | OMe | C6H13 | 3f | 55 | 1.6:1 |
94 |
| 7f | OMe | C7H15 | 3g | 51 | 1.8:1 |
90 |
| 8 | OMe | 2-ClC6H4 | 3h | 80 | 7.7:1 |
100 |
| 9 | OMe | 4-MeC6H4 | 3i | 88 | >30:1 |
100 |
| 10 | OMe | 4-ClC6H4 | 3j | 92 | 2.1:1 |
100 |
| 11 | OMe | 2-FC6H4 | 3k | 82 | 2:1 |
100 |
| 12 | OMe | 4-EtC6H4 | 3l | 87 | 6.3:1 |
100 |
| 13 | OMe | 4-NO2C6H4 | 3m | 95 | 5.8:1 |
100 |
| 14 | OMe | 3-MeO-4-AcOPh | 3n | 76 | 3:1 |
97 |
| 15 | OMe | 4-AcOC6H4 | 3o | 85 | 14.3:1 |
100 |
| 16 | OMe | 4-MeOCOC6H4 | 3p | 88 | 1.5:1 |
100 |
| 17 | OMe | 4-Ph-Ph | 3q | 83 | 4.8:1 |
100 |
| 18 | OMe | 1-Naphth | 3r | 85 | >30:1 |
100 |
| 19 | Ph | Ph | 3s | 76 | >30:1 |
100 |
| 20 | C2H5 | Ph | 3t | 94 | >30:1 |
100 |
Finally, to demonstrate the synthetic utility of the Michael adducts 3, a series of organic transformations were performed using 3a as an example (Scheme 2). Reduction of the aldehyde or reductive aminations followed by spontaneous cyclization led to synthetically valuable chiral lactone 5 and lactam 6, respectively (eqn (1)). These two classes of compounds have important biological activities1 and serve as important building blocks in organic synthesis. Moreover, the sulfone can be removed conveniently under mild conditions (eqn (2)). Protection of the aldehyde moiety to corresponding acetal 3′a, and subsequent reductive removal of the 2-pyridinylsulfonyl group under the optimized conditions using activated Mg(0) in the presence of Bu4N+Br− (TBAB) in MeOH11e (see ESI‡) gave 3-substituted acetal 7, which was transformed into an aldehyde ester 8. It is noteworthy that our initial attempt to form the methoxycarbonylmethylated product under basic conditions (Mg, TBAB, MeOH) without protection of the aldehyde resulted in a significantly low yield of ca. 10%.
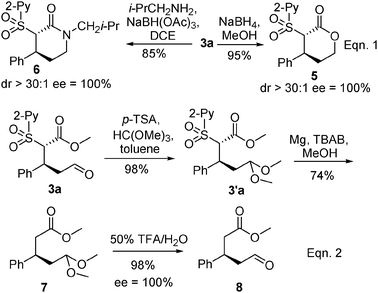 | ||
| Scheme 2 Application transformations of Michael adduct 3a. | ||
In conclusion, we have developed a new nucleophilic reagent CMPS 1a for the direct organocatalytic asymmetric Michael addition to a wide range of α,β-unsaturated aldehydes in moderate to excellent yields and with high enantioselectivities. Moreover, as demonstrated, the Michael adducts serve as versatile building blocks in a variety of transformations. The application of the process in the synthesis of biologically interesting molecules is being pursued in our laboratories.
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (Grants 90813005, 21002028 and 20902022, J. Li), National S&T Major Project, China (Grant 2011ZX09102-005-02, J. Li), the 111 Project (Grant B07023, J. Li and W. Wang), the Fundamental Research Funds for the Central Universities (J. Li) and East China University of Science & Technology (W. Wang).
Notes and references
- For reviews, see: (a) J. D. Connolly and R. A. Hill, Dictionary of Terpenoids, Chapman and Hall, London, 1991, vol. 1, p. 476 Search PubMed; (b) M. Kidwai, P. Sapra and K. R. Bhuskan, Curr. Med. Chem., 1999, 6, 195 CAS; (c) K. Bush, M. Macielag and M. Weidner-Wells, Curr. Opin. Microbiol., 2004, 7, 466 CrossRef CAS.
- To our knowledge, only two examples can be found in the literature in asymmetric preparation: the use of catalytic asymmetric Diels–Alder reactions: (a) D. A. Evans, J. S. Johnson and E. J. Olhava, J. Am. Chem. Soc., 2000, 122, 1635 CrossRef CAS; (b) D. A. Evans and J. S. Johnson, J. Am. Chem. Soc., 1998, 120, 4895 CrossRef CAS; Chiral auxiliary: (c) D. Enders and B. E. M. Rendenbach, Chem. Ber., 1987, 120, 1223 CAS.
- For recent reviews on enantioselective Michael addition reactions, see: (a) Catalytic asymmetric conjugate reactions, ed. A. Cordova, Wiley-VCH, Weinheim, Germany, 2010 Search PubMed; (b) J. L. Vicario, E. Reyes, D. Dadia and L. Carrillo, in Catalytic asymmetric conjugate reactions, ed. A. Cordova, Wiley-VCH, Weinheim, Germany, 2010, ch. 6, p. 619 Search PubMed.
- (a) S. Brandau, A. Landa, J. Franzén, M. Marigo and K. A. Jørgensen, Angew. Chem., Int. Ed., 2006, 45, 4305 CrossRef CAS; (b) F. Wu, R. Hong, J. Khan, X. Liu and L. Deng, Angew. Chem., Int. Ed., 2006, 45, 4301 CrossRef CAS; (c) Y. Wang, P. Li, X. Liang and J. Ye, Adv. Synth. Catal., 2008, 350, 1383 CrossRef CAS; (d) A. Ma, S. Zhu and D. Ma, Tetrahedron Lett., 2008, 49, 3075 CrossRef CAS; (e) Y. Hayashi, M. Toyoshima, H. Gotoh and H. Ishikawa, Org. Lett., 2009, 11, 45 CrossRef CAS; (f) A.-N. Alba, X. Companyo, A. Moyano and R. Rios, Chem.–Eur. J., 2009, 15, 7035 CrossRef CAS; (g) I. Fleischer and P. A. Ivana, Chem.–Eur. J., 2010, 16, 95 CrossRef CAS.
- A. Carlone, M. Marigo, C. North, A. Landa and K. A. Jørgensen, Chem. Commun., 2006, 4928 RSC.
- (a) C. Zhong, Y. Chen, L. Petersen, N. G. Akhmedov and X. Shi, Angew. Chem., Int. Ed., 2009, 48, 1279 CrossRef CAS; (b) S. K. Ghosh, Z. Zheng and B. Ni, Adv. Synth. Catal., 2010, 352, 2378 CrossRef CAS; (c) M. Kamlar, N. Bravo, A.-N. R. Alba, S. Hybelbauerova, I. Cisarova, J. Vesely, A. Moyano and R. Rios, Eur. J. Org. Chem., 2010, 5464 CrossRef CAS.
- (a) T. Furukawa, Y. Goto, J. Kawazoe, E. Tokunaga, S. Nakamura, Y. Yang, H. Du, A. Kakehi, M. Shiro and N. Shibata, Angew. Chem., Int. Ed., 2010, 49, 1642 CAS; (b) A.-N. Alba, X. Companyo, A. Moyano and R. Rios, Chem.–Eur. J., 2009, 15, 7035 CrossRef CAS; (c) A.-N. Alba, X. Companyo, A. Moyano and R. Rios, Chem.–Eur. J., 2009, 15, 11095 CrossRef CAS.
- D. A. Alonso, S. Kitagaki, N. Utsumi and C. F. Barbas III, Angew. Chem., Int. Ed., 2008, 47, 4588 CrossRef CAS.
- The studies from our group: (a) J. Wang, H. Li, L.-S. Zu, W. Jiang, H.-X. Xie, W.-H. Duan and W. Wang, J. Am. Chem. Soc., 2006, 128, 12652 CrossRef CAS; (b) L.-S. Zu, H.-X. Xie, H. Li, J. Wang and W. Wang, Adv. Synth. Catal., 2007, 349, 2660 CrossRef CAS; (c) L. Zu, H. Xie, H. Li, J. Wang, X. Yu and W. Wang, Chem.–Eur. J., 2008, 14, 6333 CrossRef CAS; (d) S.-L. Zhang, J. Li, S.-H. Zhao and W. Wang, Tetrahedron Lett., 2010, 51, 1766 CrossRef CAS; (e) H. Li, Y.-F. Ji, J. Li, S.-L. Zhang, C.-G. Yu and W. Wang, Sci. China Chem., 2010, 53, 135 Search PubMed; (f) S.-L. Zhang, Y.-N. Zhang, Y.-F. Ji, H. Li and W. Wang, Chem. Commun., 2009, 4886 RSC; (g) S.-L. Zhang, H.-X. Xie, J. Zhu, H. Li, X.-S. Zhang, J. Li and W. Wang, Nat. Commun., 2011, 2, 1214 Search PubMed.
- For recent reviews on sulfone chemistry, see: (a) A.-N. R. Alba, X. Companyo and R. Rios, Chem. Soc. Rev., 2010, 39, 2018 RSC; (b) M. Nielsen, C. B. Jacobsen and K. A. Jørgensen, Angew. Chem., Int. Ed., 2010, 49, 2668 CAS; (c) A. El-Awa, M. N. Noshi, X. M. Jourdin and P. L. Fuchs, Chem. Rev., 2009, 109, 2315 CrossRef CAS.
- For selected examples of sulfones as nucleophiles in conjugate additions: (a) L. Jiang, Q. Lei, X. Huang, H.-L. Cui, X. Zhou and Y.-C. Chen, Chem.–Eur. J., 2011, 17, 9489 CrossRef CAS; (b) M. G. Brant, C. M. Bromba and J. E. Wulff, J. Org. Chem., 2010, 75, 6312 CrossRef CAS; (c) N. Holub, H. Jiang, M. W. Paixão, C. Tiberi and K. A. Jørgensen, Chem.–Eur. J., 2010, 16, 4337 CrossRef CAS; (d) J. L. García Ruano, V. Marcos and J. Alemán, Chem. Commun., 2009, 4435 RSC; (e) M. Nielsen, C. B. Jacobsen, M. W. Paixão, N. Holub and K. A. Jørgensen, J. Am. Chem. Soc., 2009, 131, 10581 CrossRef CAS.
- For selected examples of sulfones as electrophiles in conjugate additions: (a) H. Li, J. Song, X. Liu and L. Deng, J. Am. Chem. Soc., 2005, 127, 8948 CrossRef CAS; (b) A. Quintard and A. Alexakis, Chem.–Eur. J., 2009, 15, 11109 CrossRef CAS; (c) Q. Zhu and Y. Lu, Org. Lett., 2009, 11, 1721 CrossRef CAS; (d) A. Landa, M. Maestro, C. Masdeu, A. Puente, S. Vera, M. Oiarbide and C. Palomo, Chem.–Eur. J., 2009, 15, 1562 CrossRef CAS; (e) A.-N. Alba, X. Companyo, G. Valero, A. Moyano and R. Rios, Chem.–Eur. J., 2010, 16, 5354 CAS; (f) E. Rodrigo, S. Morales, S. Duce, J. L. G. Ruano and M. B. Cid, Chem. Commun., 2011, 47, 11267 RSC.
- J. B. Baudin, G. Hareau and S. A. Julia, Tetrahedron Lett., 1991, 32, 1175 CrossRef CAS.
- For a review of diaryl prolinol ethers catalysis, see: A. Mielgo and C. Palomo, Chem.–Asian J., 2008, 3, 922 Search PubMed.
- We observed 24% yield of byproduct generated from 1a and hex-2-enal. (a) N. A. Magomedov, P. L. Ruggiero and Y.-C. Tang, Org. Lett., 2004, 6, 3373 CrossRef CAS; (b) J.-M. Zhang, E. A. Polishchuk, J. Chen and M. A. Ciufolini, J. Org. Chem., 2009, 74, 9140 CrossRef CAS.
- CCDC 844031 (3j).
Footnotes |
| † This article is part of the joint ChemComm–Organic & Biomolecular Chemistry ‘Organocatalysis’ web themed issue. |
| ‡ Electronic supplementary information (ESI) available: Experimental details and spectroscopic data for the compounds 3a–3t, 5–8. CCDC 844031. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1cc15714k |
| This journal is © The Royal Society of Chemistry 2012 |