Dark uncertainty
Analytical Methods Committee, AMCTB No 53
First published on 13th July 2012
Abstract
There is now abundant evidence that we analytical chemists are tending to underestimate the uncertainty of our measurements. There are two main underlying reasons for this. One reason is technical: it is easy to overlook important contributions to uncertainty, so the models used to estimate uncertainty may be incomplete. The second reason may be psychological: there may be an unconscious selection bias in the information we use to assess uncertainty. What should we do about this missing, ‘dark’, uncertainty?
A recent meta-analysis1 has reviewed available studies of reported uncertainties in inter-laboratory exercises and examined additional examples of metrology comparisons in analytical chemistry. Although the number of such studies is modest, all those reviewed show evidence that uncertainty is more often underestimated than overestimated – that is, differences among laboratories are usually greater than the reported uncertainties would suggest. As an example of this common occurrence, Fig. 1 shows the results, with their reported uncertainties, for lead in tuna, produced by participants in IMEP 20. (IMEP is the International Measurement Evaluation Programme organised by the European Institute for Reference Materials and Measurements (IRMM), Geel, Belgium). Fig. 2 shows the observed distribution of the sorted results, together with bootstrapped estimates of the expected distribution had the uncertainty estimates been correct. On the basis of the reported uncertainties, the between-laboratory standard deviation should be 0.031 ppm: the observed robust value (that is, outliers discounted) was 0.122 ppm.

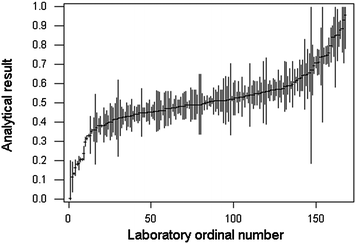 | ||
| Fig. 1 Ordered results for Pb in tuna (mg kg−1), with reported expanded uncertainties (vertical lines), from laboratories participating in IMEP20. Redrawn with permission from data published by IRMM. | ||
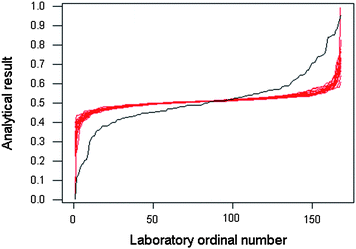 | ||
| Fig. 2 Distribution of ordered reported results for Pb in tuna (mg kg−1) (black line) and bootstrapped expected distributions (red lines) based on the reported uncertainties (same data as Fig. 1). The width of the bundle of red lines gives an idea of the uncertainty of the position of the expected distribution. | ||
This kind of occurrence is neither especially novel nor peculiar to analytical chemistry, as we can see from the classic 1972 paper by Youden2 on estimates of the velocity of light. But why now, two decades after the publication of the Guide to the Expression of Uncertainty in measurement (“the GUM”),3 should this still happen?

GUM
The GUM provided three things. First, it provided some basic concepts, such as the concept of measurement uncertainty itself as a summary figure that includes all possible effects, random or systematic, on a result. Second, it provided a set of principles for estimating uncertainty: the idea that uncertainty arises from multiple sources; that for combination, uncertainties should all be expressed in the form of standard deviations; that these ‘standard uncertainties’ should be combined using the established rules for combining variances; and the then quite radical idea that uncertainties for both random and systematic effects should be treated identically, no matter whether they were estimated from statistical analysis (“Type A”) or from other sources (“Type B”) such as calibration certificates, manufacturer specifications or professional judgement. Finally, the GUM provided a particular approach to the combination of uncertainties, based on an equation (the ‘measurement model’) that was assumed to include all known significant effects on the measurement result. This particular methodology has been described as the ‘bottom up’ approach because of its focus on building up an uncertainty budget from individual parts.Building on GUM
Since the publication of the GUM, other approaches have become available that respect the same principles but use alternative combination methods or simpler models. In particular, the second edition of the Eurachem Guide4 described a general approach using available data, including the use of in-house validation data or inter-laboratory reproducibility data as well as allowing the ‘bottom up’ approach, to evaluate uncertainty. This guide uses a combination of cause and effect analysis and a ‘reconciliation’ step to assess whether the data available are sufficient. A recent ISO Standard, ISO 21748,5 gives more detail for the use of reproducibility data, based on a simplified model equation. Approaches based on method performance data are often called ‘top down’ approaches in contrast to the reductionist ‘bottom up’ approach above. A Eurolab guide and a NordTest guide have also added some approaches for the use of proficiency testing data.6,7 Following these guides should provide analysts with comprehensive, and sometimes conservative (that is, large), estimates of uncertainty. Yet the evidence is that many, if not most, laboratories still underestimate uncertainties. So where should we look for ‘missing’ uncertainty? And what can we do about it?‘Cause and effect’ analysis
Cause and effect analysis is used to identify possible sources of uncertainty. It is usually documented in the form of an Ishikawa or ‘fishbone’ diagram, showing the different factors affecting the result. The Eurachem guide suggests that the process begin with the parameters in the equation used to calculate the measurement result. It then suggests examining each of these to identify operations or other input quantities that can affect the measurement result. As an example, consider the example of a simple pesticide residue analysis of a foodstuff measured by single-point calibration. We weigh the foodstuff to give a mass m. We extract the foodstuff with an organic solvent, clean up with, for example, solid phase extraction, and make up the resulting extract to a volume v. We make up a standard solution of known concentration c. Then we use chromatography to determine peak intensities Ix and Istd for the test material extract and standard solution respectively, and calculate the mass fraction x of pesticide in the original foodstuff using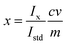
From the simple equation above, the initial components of uncertainty might stem from:
• The concentration c of the analyte in the calibrators;
• The volume v of the extract.
• The peak area ratio.
• The mass of sample taken for analysis.
These primary contributions can be further broken down into secondary contributions. For example, the concentration of analyte in the calibrator would be affected by:
• Uncertainty in the purity of the chemical standard;
• Gravimetric and volumetric uncertainties.
The process is continued until the scientist is convinced that all relevant effects are included. For example, the volumes will be affected by precision, calibration and, for completeness, temperature effects. The Eurachem guide suggests further refinement of the diagram to resolve any apparent duplication and to group related effects. Often, consideration of the analytical process identifies new factors (such as extraction efficiency) which lead to additional ‘branches’ in the diagram.
In principle, each item in the diagram is a possible contribution to uncertainty and a standard uncertainty allocated to each. This corresponds exactly to the detailed GUM approach. However, the Eurachem guide indicates that is often possible to assess the uncertainty for groups of related effects. For example, a good estimate of long term precision includes variation from a large number of effects, particularly random effects, and can reduce or eliminate the need for individual assessment of many terms. In particular, inter-laboratory reproducibility conditions allow variation (within permitted ranges) of nearly all effects on the result; the Eurachem guide therefore suggests that the reproducibility standard deviation is a good basis for an initial estimate of uncertainty (although it does add that an inter-laboratory study does not include all effects, particularly parts of sample preparation). This implies a range of possible approaches, from detailed assessment of every individual contribution through to the use of a much simpler (if less informative) summary figure of performance. And indeed both approaches are widely used in practice. But does either of these extremes guarantee an accurately estimated uncertainty?

Two schools of thought
The measurement community often seems polarised towards one or other of two extreme points of view.• The ‘bottom-uppers’ or ‘splitters’ believe that the deconstruction procedure should be exhaustive, continued to provide a complicated complete ‘model’ of the procedure. ‘Splitters’ assert (correctly in most instances) that reproducibility standard deviation tends to underestimate standard uncertainty because inter alia the effects of method bias are not accounted for. The issue of traceability is also raised: how is the outcome traceable to the SI?
• The ‘top-downers’ or ‘lumpers’ believe that deconstruction should be terminated at the earliest possible point that gives rise to a reasonable estimate of uncertainty. The extreme version of the ‘lumper’ approach is simply to use reproducibility standard deviation (obtained by replication of the entire procedure in different laboratories) as their estimate of standard uncertainty. ‘Lumpers’ take the view (again correctly in most instances) that analytical procedures involve chemical interactions so numerous and complex that it is usually impossible to build a comprehensive model. There are both hidden influences on the result and unknown interactions between overt influences. The outcome is ‘dark uncertainty’,1 present in the result of the measurement but not visible in the uncertainty budget. However, all of the effects, known and unknown (but excluding method bias), will be taken into account in reproducibility precision, because each laboratory using the procedure will explore the variable space differently and more-or-less at random. Because of this, dark uncertainty will be manifest in the reproducibility standard deviation, even though we do not know its source.
Advocates of both of these views, then, claim that the alternative method tends to under-estimate uncertainty. But these contentions are open to testing. A recent study of chemical measurement8 has found a strong tendency for reproducibility standard deviation to be greater than an estimate based on a splitter approach, by a factor of about 1.5–2. And reproducibility standard deviation itself is potentially too small: it does not account for method bias. Dark uncertainty seems to be not only ubiquitous but almost inevitable in chemical measurement. So what should the analyst do?
Checking the reliability of uncertainty estimates
An obvious place to start is to check whether uncertainty estimates are realistic. This is covered in another AMC Brief,9 so we will not discuss it in detail here. But as a simple rule of thumb, an uncertainty estimate much better than typical reproducibility standard deviations sR found for relevant methods and test materials should be reviewed as suspect. Where no relevant studies are available, relevant guidance (often regulatory) on acceptable performance may be a useful guide. And in the food analysis sector, Horwitz's compilations have demonstrated a strong general tendency for reproducibility standard deviation to be about twice the associated repeatability standard deviation sr (that is, sR ≈ 2sr) so a general tendency for uncertainty estimates in a laboratory to be less than 2sr should be regarded as suspect. Where we should look, once we have identified a potential problem, depends on the approach we have taken for our uncertainty estimate. The GUM assumes that we have an equation that describes, quantitatively, all known, significant effects on the result. This is one obvious place to look for missing uncertainties.‘Bottom-up’ analysis from the model equation
In principle, we can apply the GUM approach to the equation in the pesticide example above. A cursory examination might suggest that chromatographic peak areas can be estimated with a (relative) standard uncertainty of about 1%, that masses and volumes can be determined with uncertainty near 0.1% and that the stock solution uncertainty (which depends on further weighings and volumetric operations) could be known with relative uncertainty well under 1%. Combining these in the usual way gives a relative standard uncertainty of the order of 1.5–2%. We might see a repeatability relative standard deviation of 5–15% on spiked test materials, so the estimated relative uncertainty could be, perhaps, 10%.This may be a fair summary of the combination of known calibration uncertainties and observed repeatability – and indeed confirms very nicely that we need take no further care over our instrument and glassware calibrations, which are contributing very little to the uncertainty. But it will not take a working analyst long to work out that the model used is woefully incomplete. Organic trace analysis is critically dependent on efficient extraction and minimal loss.
Shortcomings can cause very large biases – but neither appears in the ‘model’ above. Nor is it simple to incorporate them; although we can easily add a nominal ‘recovery correction’ factor to the above model, with a large uncertainty, we still need to characterise that uncertainty. In practice we can rarely characterise extraction processes sufficiently well for a given test material, and losses from oxidation, evaporation, SPE cartridge retention, and photochemical and chemical degradation are very hard to characterise in any quantitative way.
This, then, is one place to look for missing uncertainties. The principal weakness of the ‘bottom up’ approach for routine testing is that the largest effects are often too poorly characterised to include in a quantitative model, and can at best be limited by careful procedure. A slightly more subtle problem is that no model can include effects the scientist is not yet aware of, making extensive experience and training very important if this approach is used.
“Top-down” analysis
Top-down estimates of uncertainty use method performance data; typically an estimate of precision, an estimate of bias plus perhaps some additional allowances. The critical questions are then “which estimate of precision?” and “of which measurement?”Precision can be estimated from any set of repeated observations, from re-presentation of an extract to an instrument, through repetition of the complete measurement with no changes in calibrations, operator or equipment, to repetition by different laboratories. But the estimates of precision we get under these different conditions are very different, and we need to choose the right one. In one study of uncertainties reported in proficiency tests, it was found that those laboratories using repeatability standard deviation as the basis for their reported uncertainty were by far the most likely to show errors much larger than their reported uncertainty would suggest.8 Repeatability standard deviations do not tease out all the hidden, and often large, effects. The lesson is clear: repeatability standard deviations alone are insufficient for measurement uncertainty estimation and we must use conditions that encompass as large a range of effects as possible.
Bias estimates used for uncertainty estimation have been less studied. However, we do know that if we measure recovery on a single simple material, we are likely to get rather more favourable answers than by looking at a range of different matrices, and while a poor spike recovery is a reliable sign of a problem, a good spike recovery could simply reflect insufficient equilibration or a less strongly bound material, yet another hidden uncertainty. We must choose our bias studies from the hard cases as well as the easy cases to get realistic uncertainty estimates.

Selection bias and fitness for purpose
A contribution to underestimation of uncertainty is our tendency to use specially prepared test materials for estimating precision. This can give rise to an underestimated precision and a comfortable feeling that the method is more accurate than it actually is. Often the materials used in estimating precision are finely ground and well-mixed control materials, or even certified reference materials. Such materials can give rise to better precision estimates than achieved with routine test materials, by virtue of being closer to homogeneity and therefore more effectively decomposed by the chemical operations preceding instrumental measurement. That is a reasonable strategy for studying the method per se. But in uncertainty estimation we should not be interested in the method per se but in the performance of the whole analytical system, the combination of the method and the laboratory samples prepared in the routine way. We should use for that purpose test materials that are typical of those encountered under conditions of routine analysis.The subconscious tendency to prefer results that look good is natural enough, but is partly founded on a ‘target culture’ derived from training. Our early attempts at chemical analysis are unskilful and we are trained to develop skill by trying for the smallest possible uncertainty. This strategy is sensible as far as it goes, but has an unfortunate side effect. We are led to feel uncomfortable if we do not achieve this low uncertainty. But ultimately we need judgement as well as skill. Fitness for purpose demands an uncertainty that is optimal for the customer in terms of overall cost, not the smallest possible. The overall cost is the cost of the measurement per se, plus the cost of a mistaken decision based on the result (and its probability). Lower uncertainty means a higher measurement cost but a lower chance of a mistake. We have to achieve the best balance between these costs. There should be no comfort in demonstrating the achievement of an unnecessarily small uncertainty.
There is also the commercial aspect: we may be worried about offering an optimal uncertainty in case our competitors are offering an unnecessarily (and often unrealistically) small one. This is a serious problem: customers simply complying with an item in a quality manual will tend to select price-for-price the laboratory that seems to offer the lowest uncertainty. The problem can be alleviated only by education of the customer, a formidable task but one that should be attempted as part of an analyst's professional activities. As well as explaining the causes and outcomes of unrealistically small uncertainty estimates, laboratories tendering for contracts should strongly encourage potential customers (i) to require uncertainty specifications from all their competitors and (ii) to apply quality control measures on the contracted-out analysis to ensure that the specification is being met.
This Technical Brief was drafted for the AMC by M Thompson and S L R Ellison.

References
- M. Thompson and S. L. R. Ellison, Accredit. Qual. Assur., 2011, 16, 483–487 CrossRef.
- J. Youden, Enduring values, Technometrics, 1972, 14, 1–11 Search PubMed.
- ISO/IEC Guide 98:1993, Guide to the expression of uncertainty in measurement (GUM), International Standards Organization, Geneva, 1993. also Available Free as JCGM 100:2008; see http://www.bipm.org/en/publications/guides/gum.html Search PubMed.
- Quantifying Uncertainty in Analytical Measurement, Eurachem/CITAC Guide, ed. A. Williams, S. L. R. Ellison and M. Roesslein, 2nd edn, 2000, available from the Eurachem secretariat and website (http://www.eurachem.com/) and (hard copy) LGC Ltd, London (ISBN 0-948926-15-5) Search PubMed.
- ISO 21748:2010, Guidance for the Use of Repeatability, Reproducibility and Trueness Estimates in Measurement Uncertainty Estimation, International Standards Organization, Geneva, 2010 Search PubMed.
- NordTest Technical Report 537, Handbook for Calculation of Measurement Uncertainty in Environmental Laboratories, See http://www.nordtest.info Search PubMed.
- Eurolab Technical Report No. 1/2007, Measurement Uncertainty Revisited: Alternative Approaches to Uncertainty Evaluation, Available at http://www.eurolab.org Search PubMed.
- S. L. R. Ellison and K. Mathieson, Accredit. Qual. Assur., 2008, 13, 231–238 CrossRef CAS.
- AMC Technical Briefs No 15 (Dec 2003) “Is my uncertainty estimate realistic?”.
| This journal is © The Royal Society of Chemistry 2012 |