Proteinaceous binders identification in the works of art using ion-pairing free reversed-phase liquid chromatography coupled with tandem mass spectrometry†‡
Bartłomiej
Witkowski
,
Magdalena
Biesaga
and
Tomasz
Gierczak
*
Faculty of Chemistry, Warsaw University, al. Zwirki i Wigury 101, 02-089 Warsaw, Poland. E-mail: gierczak@chem.uw.edu.pl
First published on 4th April 2012
Abstract
A simple, fast and reliable procedure for the proteinaceous binders identification in the works of art samples is presented. The procedure consisted of ammonia extraction in order to suppress pigment interferences, acidic hydrolysis and quantification of underivatized amino acids using reversed phase liquid chromatography coupled with electrospray tandem mass spectrometry (RP-LC–ESI-MS/MS). Fourteen underivatized amino acids were quantified without the addition of ion-pairing agents (IPA) using the multiple reaction monitoring (MRM) mode. The chromatographic separation was optimized by testing three C18 columns and three different eluent compositions. The optimal chromatographic resolution and the ionization efficiency were achieved with Symmetry C18 column and the eluent consisting of water with methanol. The amino acids composition of the proteins commonly found in the paint binding media, eggs, casein and animal glues, was determined. The procedure was tested using a set of naturally aged samples. Calculated detection and quantification limits indicated that the method is suitable for the analysis of protein binders in the paint micro-samples. Animal glues, casein and eggs were identified in the samples from 18th and 19th century paintings by Jacek Malczewski, while eggs and casein were detected in the mural painting samples from the 13th century UNESCO-listed church of Yemrehanna Krestos. The LC/MS/MS based method of the protein binders identification described here can be used as an alternative for the approach based on the gas chromatography coupled with mass spectrometry (GC/MS).
1. Introduction
Protein binders have been used by artists since ancient times, and are listed in the numerous different recipes. Chicken eggs, collagen and casein are proteins commonly found in painting binding media. Egg yolk and whole egg are used as binders in the tempera paintings, forming elastic and resistant films upon drying; egg white is used mostly as a varnish. Animal collagen glues are prepared by boiling the soft tissues of animals or fish, and are sometimes used as paint binders, but are mainly found in ground layers, mixed with chalk or gypsum. Casein tempera was a less popular alternative for the egg tempera, used mostly in mural paintings, while strong casein glue could be sometimes found in painting grounds and fixatives.1–4 Conservation and restoration of the historical paintings require the chemical composition characterization of the organic paint media. Modern analytical chemistry is capable of providing this information.A variety of techniques have been used to study protein binders in the artworks. The most basic microchemical test involves studying the physical properties of the binding medium like solubility or melting point. Immunological methods are more recent variants of the microchemical tests, especially suitable for protein binders identification.5–10 Spectroscopic techniques including UV-Vis spectroscopy,11 Fourier transform infrared spectroscopy (FTIR)12–16 as well as Raman spectroscopy (IR)17 were used to characterize protein binders. Many of the spectroscopic techniques have the advantage of being completely non-invasive. Direct infusion ESI-MS has been also applied to analyze protein binders.18 Identification of proteins in the works of art and archeological samples has been also performed with proteomics.19–24 MALDI together with PCA analysis was described as a method of proteinaceous binders identification.25 MALDI-TOF has been used to characterize egg and oil-based binders by monitoring specific oxidation by-products.26 The detailed description of the modern analytical methods for studying organic substances in the works of art can be found in a review by Doménech-Carbó.27
In contrast to spectroscopic techniques, chromatography is capable of separating compounds of interest from complex matrices, and can provide more detailed information about the sample. Gas chromatography coupled with mass spectrometry (GC/MS) has been most frequently applied to analyze amino acid derivatives and identify protein binders.1,28–43 Aside from the classical derivatization approach, pyrolysis interfaced GC/MS was also used to study protein binding media in the works of art.1,40,44–46 Chromatographic methods of characterizing proteinaceous binders were summarized by Colombini et al.47,48
High performance liquid chromatography (HPLC) appears to be the most suitable for amino acids analysis. Unfortunately, the analysis of amino acids by reversed phase liquid chromatography (RP-LC) is problematic due to poor retention and low UV-Vis absorption, therefore derivatization is often necessary.49–56 The separation of underivatized amino acids by reversed phase liquid chromatography (RP-LC) requires addition of ion-pairing agents (IPA) like trifluoroacetic acid (TFA).57 Eluents used in RP-LC are highly compatible with MS detection; therefore reverse phase ion-pairing LC coupled with electrospray ionization mass spectrometry (LC-ESI/MS) has become a very popular technique in amino acids analysis.58–65 However, it has been proven that ion pairing agents cause significant ionization suppression in ESI. Strong ion-pairing with TFA anions masks the analyte and limits the emission of protonated molecules into the gas phase. Addition of TFA to the eluent can also produce unstable spray due to high conductivity and surface tension of the droplets.66–68 To minimize signal suppression it is necessary to use different ionization techniques69 or post-column additives.67,70,71 Therefore the development of a simple, non-time consuming, derivatization and ion-pairing free proteinaceous binders identification method using liquid chromatography coupled with electrospray ionization tandem mass spectrometry (LC-ESI/MS/MS) would be of great value. Only recently, when the experiments described here were under way, Zangrando et al.72 described the procedure for identification of protein binders using an ion-pairing and derivatization free ESI-LC/MS method. The described method involved the use of hydrophilic interaction (HILIC) column and multiple reaction monitoring (MRM) mode to quantify partially resolved amino acids after a 55 min gradient program. Also, special attention was paid to resolve isobaric amino acids that cannot be distinguished by MS alone. However to date, no work describing a fast method of proteinaceous binders identification using the C18 column and tandem MS detection in analysis of underivatized amino acids without the use of IPA has been published.
Several methods for obtaining free amino acids mixture from the proteinaceous binding materials were developed. Acidic hydrolysis with 6 M HCl in the gas or liquid phase at 110 °C for 24 h is most commonly used. Some variations of this method include shorter reaction times (e.g. 5 h), additives to protect less stable amino acids or applying microwave radiation. Enzymatic hydrolysis enables analysis of less stable amino acids, but long reaction times and problems in achieving complete digestion are significant drawbacks.1,42,47,73–76
Before hydrolysis, sample clean-up is often necessary to suppress matrix interferences. Inorganic pigments were proven to cause significant problems in paint binding media analysis. Proposed clean-up procedures include removal of inorganic salts by cation exchangers, use of complexing agents like EDTA, or extraction with NaOH, NH3 and H2O.1,28,29,39,41,47
In spite of the growing contribution of HPLC-based methods to the studies of organic binders in the works of art, at the present time GC/MS is still the major method used in this field. Amino acids analyzed by GC have to be transformed into corresponding derivatives to enhance volatility and thermal stability. Aside from being laborious and time consuming, all derivatization procedures possess some drawbacks. For instance: the intolerance to even small amounts of water in the case of silylating agents. Derivatization with chloroformates can be performed in the aqueous solutions; however, limited reactivity of those reagents can produce problems when analyzing some amino acids.30,34,77,78 Derivatives have to be extracted in chloroform and dried; pH has to be controlled in order to achieve a high yield. Complex sample processing in the most popular derivatization procedures increases the probability of sample contamination and limits the number of amino acids that can be analyzed. Frequently, in order to obtain complete information about the binding medium, it is necessary to identify multiple types of organic materials used in paint preparation.1,48 Despite the fact that LC/MS is suitable for the analysis of e.g. fatty79 and resin acids,80,81 up to date, no papers describing LC-MS-based procedures for simultaneous identifications of different types of organic binders have been published. On the other hand, a number of GC/MS procedures for simultaneous identification of the different organic binders were introduced.31,33,36,43
Since the applications of LC/MS in the field of the works of art analysis are still under development, new protocols for identification of the organic binders need to be introduced. The main aim of this work was to prepare a fast and robust analytical method for the identification of the proteinaceous binding materials in the works of art. The novel method proposed here consists of the effective acidic hydrolysis of the protein-based binders after the extraction of the proteinaceous material with ammonia. Analysis of the amino acids was performed using the derivatization and ion-pairing free LC-ESI/MS/MS method with C18 column. The analytical method developed in this paper was tested on a set of model paint samples. Afterwards, the protein binders in the mural painting samples from the UNESCO-listed Ethiopian church of Yemrehanna Krestos, and samples from Jacek Malczewski paintings were investigated.
2. Experimental
2.1. Materials and methods
Amino acid standards: alanine (Ala), arginine (Arg), histidine (His), hydroxyproline (Hyp), isoleucine (Ile), leucine (Leu), lysine (Lys), methionine (Met), phenylalanine (Phe), proline (Pro), serine (Ser), threonine (Thr), tyrosine (Tyr), valine (Val) (all of purity ≥98%), as well as LC-MS grade methanol (≥99.9%) and ammonium acetate (≥99.0%), were purchased from Sigma-Aldrich (Schnelldorf, Germany). Ultrapure water for LC-MS was prepared using the Direct-Q 3 Ultrapure Water System (Millipore). Formic acid (≥98%) was obtained from Fluka, Sigma-Aldrich (Schnelldorf, Germany). PTFE membrane and syringe filters (pore size 0.45 μm) were obtained from Supelco and Sigma-Aldrich (Schnelldorf, Germany). Hydrochloric acid (≥99.0%) and 25% ammonia solution in water (≥99.0%) were obtained from POCh (Gliwice, Poland). Materials used by art conservators: rabbit glue, fish glue, collagen, casein and naturally aged model paint samples were kindly donated by the conservation laboratory of National Museum in Warsaw. Chicken eggs were obtained from the market.2.2. Model paint samples and reference materials
Rabbit glue, fish glue, collagen, casein and chicken eggs were used as reference materials. Egg white, egg yolk and mixed whole egg were spread on a glass and left to dry for a month at room temperature; other reference materials were used without any further preparations. The procedure was tested on a set of naturally aged samples: ground layer containing animal glue (M1), casein tempera (M2) and egg yolk tempera (M3). Sample M4 consisting of drying oil and resin was used as a method blank. Compositions of the model paint samples as well as aging times are provided in Table 1. All model temperas were prepared according to historical recipes and stored in dark and dry conditions in the conservation laboratory of National Museum in Warsaw before analysis.| Model paint sample | Ratios of the components (w/w) |
|---|---|
| Sample of the painting ground layer (M1) | Skin glue (5% solution in water): 2 |
| Chalk: 1.5 | |
| The sample was naturally aged for 30 years | |
| Casein tempera (M2) | Casein: 1 |
| Linseed oil: 0.5 | |
| Damara resin: 0.2 | |
| Ammonia solution in water (8%): 2.5 | |
| The sample was naturally aged for16 years | |
| Egg yolk tempera on ground layer containing animal glue (M3) | Egg yolk was the only organic component of the paint layer |
| Ground layer was prepared as in the sample M1 | |
| The sample was naturally aged for 30 years | |
| Oil paint (M4)—used as protein blank sample | Linseed oil was the only organic component of the paint layer |
| Damara resin was used as a varnish | |
| The sample was naturally aged for16 years |
2.3. Samples from the works of art
A total of 9 mural painting samples from the Ethiopian church of Yemrehanna Krestos were taken by the archeological team expedition in 2007 in order to examine the paintings inside the building and prepare a report for UNESCO. The building is dated as 13th century, and it is on the World Heritage List, along with 10 other so-called rock-hewn churches. Samples were delivered as large pieces of plaster covered with a thin layer of paint on them; paint was carefully scraped off and about 1 mg was analyzed. A set of 30 samples from paint and ground layers were analyzed. Samples were taken from 18 paintings by Jacek Malczewski, dated between 1884 and 1926, from the collection of National Museum in Warsaw (Poland). A very small amount of paint and ground layers (≤0.5 mg) were sampled by art conservators as a part of restoration works and delivered for analysis.2.4. Statistical analysis
The cluster analysis was used to identify the proteinaceous binding materials in samples. A raw data matrix was constructed by assigning every sample its amino acids composition; afterwards data were standardized and the Euclidean distance matrix was calculated. Clustering was performed using the Ward method of minimizing sum of squares (SS) inside the newly formed clusters. The resulting tree diagrams consisted of similar samples grouped together according to the calculated distances. Statistical data treatment was performed using Statistica® (Statsoft, Poland).2.5. Samples preparation
Before hydrolysis, sample clean-up was performed, as described in ref. 29, in order to suppress possible interferences from inorganic pigments. Sample was extracted twice with 0.5 ml of 2.5 M NH3(aq) for 2 h at 60 °C in an ultrasonic bath. The resulting solution was centrifuged for 5 min at 4000 RPM and filtered by a 0.45 μm PTFE syringe filter. Afterwards, solution was evaporated to dryness under a gentle stream of N2 and 100 μl of 12 M HCl was added; then the hydrolysis was performed for 24 h at 110 °C. After hydrolysis, solution containing amino acids was again evaporated to dryness under a stream of N2. Residue was dissolved in 1 ml of water, filtered by a 0.45 μm PTFE syringe filter and 10 μl of this solution was subjected to the chromatographic analysis.2.6. Apparatus
All chromatographic analyses were performed using a LC20 liquid chromatograph (Shimadzu) coupled with a QTRAP 3200 mass spectrometer (Applied Biosystem/MDS SCIEX). Amino acids were separated on the Symmetry (Waters) C18 column (75 mm × 4.6 mm, 3.5 μm) kept at 30 °C. Water (eluent A) and methanol (eluent B) were used as mobile phase components. The mobile phase was delivered at a flow rate of 0.3 mL min−1 in gradient mode: 0–8 min 5% B, 10–12 min 50% B, 12.5–20 min 5% B. Other columns tested were: Zorbax (Agilent) C18 (50 mm × 4.6 mm, 1.8 μm) and Atlantis (Waters) C18 (50 mm × 2.1 mm 3 μm). The mass spectrometer was operating in the multiple reaction monitoring (MRM) mode. Electrospray ionization (ESI) conditions were: capillary temperature 450 °C, curtain and auxiliary gas at 0.3 MPa, positive ionization mode, source voltage 5.5 kV. Nitrogen was used as curtain, auxiliary and collision gas. MS/MS conditions were optimized using infusion mode; standard solutions were delivered to the ion source using a Harvard Apparatus pump at a flow rate of 10 μL min−1. Initially entrance potential (EP), declustering potential (DP), and collision cell entrance potential (CEP) were optimized for the protonated parent ions. Afterwards optimal collision energy (CE) and collision cell exit potential (CXP) were determined for the collision cell.3. Results and discussion
A development of the fast, derivatization and ion-pairing free LC-ESI/MS/MS analytical method for the amino acids analysis obtained from the proteinaceous binding materials from the artworks consists of several steps. These steps will be described in the next few paragraphs: (I) the optimization of the mass spectrometer parameters before the chromatographic analysis to obtain the highest possible sensitivity, (II) the chromatographic parameters optimization to achieve the optimal chromatographic separation and sensitivity, (III) the validation of the developed procedure to evaluate accuracy of the protein binders identification in the aged samples of known composition, and (IV) the model paint samples analysis. After confirming the accurate protein binder identification in the model paint samples, the real samples from the works of art were analyzed.3.1. Method optimization
The optimal conditions for the best performance of the mass spectrometer were established. The MS conditions for all the analyzed amino acids were optimized using the infusion mode. The EP, DP, CEP, CE and CXP were swapped and the optimal values for each MRM were determined. As shown in Fig. 1, the most intense transition for Phe is 166/120 with CE = 21. A further increase in CE for Phe leads to the formation of less intense fragments at m/z = 103, 93, 91, 79, 77 and 51 Da. In order to achieve the highest possible sensitivity for all the quantified amino acids, the most intense Q1/Q3 transitions were selected for the final method. The optimal values of DP and CE for all MRM's are listed in Table 2. | ||
| Fig. 1 The collision energy optimization for Phe (m/z = 166). | ||
| Amino acid | Retention time/min | Q1/Q3 transition (m/z) | CE/V | DP/V | R 2 | LOD/μg ml−1 | LOQ/μg ml−1 |
|---|---|---|---|---|---|---|---|
| Lysine (Lys) | 2.28 | 147/84 | 23 | 21 | 0.999 | 0.4 | 1.2 |
| Arginine (Arg) | 2.41 | 175/70 | 27 | 26 | 0.986 | 0.9 | 2.4 |
| Alanine (Ala) | 2.84 | 90/44 | 8 | 11 | 0.989 | 1.7 | 4.9 |
| Serine (Ser) | 2.84 | 106/60 | 15 | 21 | 0.996 | 3.6 | 9.7 |
| Hydroxyproline (Hyp) | 2.85 | 132/68, 132/86 | 25 | 26 | 0.999 | 0.4 | 1.1 |
| Histidine (His) | 2.88 | 156/110 | 19 | 26 | 0.997 | 0.2 | 0.3 |
| Threonine (Thr) | 2.95 | 120/74 | 15 | 26 | 0.998 | 1.5 | 4.2 |
| Proline (Pro) | 3.13 | 116/70 | 21 | 36 | 0.997 | 1.0 | 2.7 |
| Valine (Val) | 3.57 | 118/72 | 37 | 26 | 0.996 | 0.6 | 1.6 |
| Methionine (Met) | 4.26 | 150/104 | 13 | 26 | 0.995 | 2.2 | 6.6 |
| Tyrosine (Tyr) | 5.18 | 182/136 | 15 | 31 | 0.999 | 0.1 | 0.2 |
| Isoleucine (Ile) | 5.25 | 132/86 | 15 | 21 | 0.997 | 0.8 | 2.0 |
| Leucine (Leu) | 5.70 | 132/86 | 15 | 21 | 0.995 | 1.3 | 3.3 |
| Phenylalanine (Phe) | 10.42 | 166/120 | 21 | 31 | 0.997 | 0.5 | 1.4 |
Usually the co-eluting amino acids can be quantified in MRM mode. However, the isobaric amino acids have to be sufficiently resolved by LC, in order to be quantified separately. Therefore, three different eluents were tested in order to achieve compromise between chromatographic separation and ionization efficiency. The deionized water, 2 mM ammonium acetate (pH = 7) and formic acid (pH = 3) were tested as the A component of eluent, while methanol was used as eluent B. Fig. 2 shows a significant decrease in the amino acids detection sensitivity in 2 mM ammonium acetate compared with water. An observed decrease in sensitivity can be explained by similar values of the gas-phase proton affinities (PA) for ammonia and amino acids.82,83 It has been proven that the gas-phase proton transfer reactions have a significant impact on the ionization process in ESI, and the solvent components with high PA can suppress ionization of the analyte molecules.66,84,85 As expected, ionization is enhanced when water is substituted with formic acid (pH = 3). However, the addition of formic acid lowers the chromatographic resolution, by decreasing the interactions between the protonated amino acids and the stationary phase. To maintain the optimal chromatographic separation of isobaric amino acids (Ile, Leu and Hyp), and the sufficient sensitivity, water (A) and methanol (B) were chosen as eluent components.
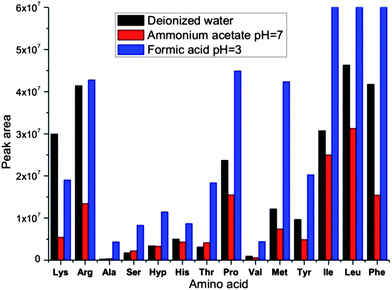 | ||
| Fig. 2 The effect of eluents composition on ionization efficiency. | ||
Three C18 columns were tested to optimize the chromatographic separation. The resolution obtained using Zorbax C18 and Atlantis C18 was insufficient, with the majority of the amino acids eluting near the void peak as shown in Fig. S1 and S2‡. The best chromatographic resolution without addition of the ion-pairing modifier was obtained using the Symmetry C18 column. Even at the optimal chromatographic conditions, no baseline separation could be obtained for two isobaric amino acids, Ile and Leu. Hyp was completely separated from Ile with RS = 2.47. As indicated by linear regression coefficients (R2) values (see Table 2) the integration of the non-separated Ile and Leu peaks was very reproducible. Other partially separated amino acids are easily determined using MRM mode as shown in Fig. S3‡; therefore there is no need for complete resolution by LC. MRM's listed in Table 2 are in very good agreement with the literature data;58,60,63,64 transition 132/86 is common for Ile, Leu and Hyp, and was selected for the quantification of those amino acids. MRM 132/68 is characteristic for Hyp and was also included in the final method.58
Gly was eluted between 2.3 and 3 min, and a very significant ionization suppression was observed for this amino acid. Gly has the lowest PA of all the amino acids, therefore it is reasonable to assume that the gas-phase proton transfer reactions with co-eluting amino acids are suppressing ionization of Gly.82,83 Very broad peaks were observed for the aspartic acid (Asp) and the glutamic acid (Glu). At the neutral pH, Asp and Glu are fully dissociated, and both contain one additional carboxylic group compared with other amino acids. Therefore interactions with C18 stationary phase are most likely very weak. Because of such quantification problems, Gly, Asp and Glu were not included in the final method.
To check the linearity of the MS detector response, a linear regression analysis of the extracted ion currents (XIC's) peak areas versus concentration of the studied compounds was measured. The linearity was determined by the square correlation coefficients of the calibration curves generated by three repeated injections of the standard solutions at concentration levels between 0.8 and 100 (μg mL−1). As shown in Table 2, for the majority of investigated amino acids the straight lines with the regression coefficients ≥0.99 were obtained. Both limits of detection (LOD's) and limits of quantification (LOQ's) were calculated by analyzing procedure blank samples. LOD's and LOQ's reported in Table 2 were calculated using mean blank signal plus 3 or 10 standard deviations of the blank. Sample M4, oil tempera, was analyzed in order to estimate the level of protein contamination in a non-proteinaceous binding medium. Concentrations of amino acids in the sample M4 did not differ significantly from the calculated LOQ's, therefore indicating a low level of protein contamination in the oil-based binding medium.
3.2. Analysis of the model paint samples and the reference materials
When identifying the protein binding media in the works of art samples, the amino acids composition of unknown sample is compared to the composition of the reference material. Egg proteins, casein and animal glues were hydrolyzed and repeatedly analyzed to calculate their mean amino acids composition. The model paint samples were analyzed to evaluate the accuracy of the procedure, and the efficiency of possible pigment interferences suppression with ammonia extraction. Since naturally aged model paint samples were available, it was possible to estimate aging effects on accuracy of protein binders identification. Description of the model samples is reported in Table 1. All samples were prepared as described in the Experimental section. As shown in Fig. 3A, the sample M2 was correctly identified as the casein tempera, and the sample M1 was assigned to the animal glues cluster. The sample M3 containing a mixed binder was assigned to the animal glues cluster, indicating that the dominating protein was the skin glue. The Euclidean distance calculated for the sample M3 from egg and the animal glues clusters was very similar, placing this sample in between those two clusters. The sample M4 (oil paint) was used as a protein blank sample as described earlier. Analysis of the model paint samples showed the correct identification of the aged proteinaceous binders in the presence of other organic materials and inorganic pigments. | ||
| Fig. 3 Tree diagrams from cluster analysis of model paint samples (A), samples from works of Jacek Malczewski (B) and mural paint samples from the church of Yemrehanna Krestos (C). | ||
3.3. Analysis of the samples from the works of art
The amino acids were detected in all of the painting's samples from the church of Yemrehanna Krestos. All samples were assigned to the egg and casein clusters, indicating the presence of those proteins in the mural painting's samples. The XIC chromatogram of one of the samples from the church of Yemrehanna Krestos is shown in Fig. 4A. Only contamination-level concentration of Hyp was detected in the mural painting's samples, confirming that animal glues were not used in preparation of the binding medium. | ||
| Fig. 4 Paint sample from the church of Yemrehanna Krestos (A) and the extracted ion chromatograms of 5 MRM pairs of sample G1 from Malczewski painting ground layer (B). | ||
19 paint layer and 11 ground layer micro-samples from Jacek Malczewski paintings were analyzed. The amino acids were detected in 6 ground layer samples, proving that no proteins were used by the artist in the paint binding media preparation. Fig. 3B shows that samples G1–G4 were assigned to animal glues cluster, and as shown in Fig. 4B, a large Hyp peak additionally confirmed the presence of the animal glue in those samples. No Hyp peak was detected in samples G5 and G6, and those samples were assigned to casein and egg clusters, indicating the presence of those two proteins. The results obtained for those samples indicate that proteins were not the main organic components in Malczewski's works, and were used only in the ground layers preparation. The relative amino acid concentrations determined for the reference materials, model paint samples and samples from the works of art are reported in Table S1‡.
An application of HPLC for analysis of organic binders in works of art analysis has been limited, due to the lack of well established procedures. Until recently, the most popular methods for the protein binders identification by HPLC involved derivatization. All of the mentioned methods have some drawbacks for instance: the instability of derivatives and the difficulties in the analysis of some amino acids.47,55,72 The method of the analysis of 14 underivatized amino acids by LC-ESI/MS, without the addition of IPA using the HILIC column, was recently published by Zangrando et al.72 The results obtained using those two columns are somewhat different. Symmetry C18 column performed better than ZIC®-HILIC column in the analysis of Met, Arg and Tyr. The linear regression coefficients for those amino acids showed good reproducibility. Asp and Glu exhibited very poor peak shape on the Symmetry C18 column in neutral pH. At the same time there were no problems in the quantification of Asp and Glu when ZIC®-HILIC column was used. ZIC®-HILIC column also allowed the baseline separation of isobaric Ile and Leu, while full resolution was not achieved with Symmetry C18 column. However, using the Symmetry C18 column the 14 amino acids analysis can be performed almost 3 times faster. This is especially important when a large number of samples have to be analyzed. Shorter analysis time will also save the expensive, high purity eluents. In this work it was shown that a good reproducibility can be obtained without the use of isotope labeled internal standards (IS). Co-eluting amino acids can be quantified separately in MRM mode with the LOD's and LOQ's values being comparable in both methods.
The developed method was successfully applied to analyze the amino acids composition in the proteinaceous binding media in the artworks. The protein binders were identified in the paintings by Jacek Malczewski and the mural painting samples from the church of Yemrehanna Krestos. Analysis of the ground layer samples from Malczewski's paintings confirmed the presence of animal glues in those samples. Mural painting samples from the church of Yemrehanna Krestos most likely contain casein and eggs. The sample clean-up procedure suppressed the matrix interferences and allowed identification of the proteinaceous material.
4. Conclusions
The identification of the protein binders in the samples from the works of art was investigated using a novel LC-ESI/MS/MS method. Fourteen underivatized amino acids were quantified using MRM mode, without addition of the potentially problematic ion-pairing agent. While using C18 column with no full chromatographic separation, isobaric amino acids were sufficiently separated in order to obtain reproducible quantification. The developed procedure was tested on a set of naturally aged paint samples of known composition. Afterwards, samples from the works of art were analyzed. Animal glues, casein and egg proteins were identified in several samples from Jacek Maczewski works. Casein and egg proteins were detected in the paint samples taken from the wall paintings in the church of Yemrehanna Krestos.When the identification of the protein is performed on the basis of their relative amino acids content, a reliable reference materials database is essential. In this work, we have investigated the average amino acid content of the three proteins most commonly encountered in the samples from the works of art. In the future studies, the larger sample size would ensure better estimation of the error margin of their average amino acids composition. Also, adding proteins less frequently encountered in the samples from the works of art, e.g. keratin or garlic should be considered.1
It is important to notice that the method presented here is based on determining the amino acids content of the protein binders. In such a case one must be careful since the degradation due to aging and environmental factors can alter the original sample composition, as well as influence the amino acids distribution.1,47 Although in most cases, proteinaceous binders can still be correctly identified on the basis of the relative amino acids content.47,86,87 The procedure described in this study produced reliable results when analyzing significantly aged proteins, however, such a methodology cannot provide any insights into the degradation processes. The investigations of the individual degradation pathways have to be performed in a different manner.26,40,86,88,89
Although this study focused on characterization of the protein-based binders, it is an important contribution to the progress in this field. The amino acids analysis method without the derivatization procedure and with the MRM type of analyte identification has a potential to become the chromatographic method of choice in the works of art analysis. In case of the procedure described in this work, the lack of the derivatization step lowers the risk of contamination, and makes the procedure significantly simpler. The presented LC-ESI/MS/MS method is faster and less laborious than GC/MS methods.
Acknowledgements
The authors would like to thank the Structural Research Laboratory (SRL) at the Department of Chemistry of University of Warsaw for making HPLC-MS measurements possible. SRL has been established with financial support from European Regional Development Found in the Sectoral Operational Programme “Improvement of the Competitiveness of Enterprises, years 2004–2005” project no.: WPK_1/1.4.3./1/2004/72/72/165/2005/U. This work was also partially financed by 501/68-BW-172101 project. This work was supported by the European Science Foundation 2007/03-LCNANOP project co-operated with the Foundation for Polish Science MPD Programme co-financed by the EU European Regional Development Fund. This work was carried out in cooperation with National Museum in Warsaw.References
- Organic Mass Spectrometry in Art and Archaeology, ed. M. P. Colombini and F. Modugno, John Wiley & Sons, 2009 Search PubMed.
- W. Ślesiński, Painting Techniques; Organic Binders, Warszawa, 1984 Search PubMed.
- J. Hopliński, Paints and Paintings Binding Medias, Ossolineum, Wrocław, 1990 Search PubMed.
- L. Losos, Panting Techniques, Cinematic and artistic publishing house, Warszawa, 1991 Search PubMed.
- G. Sciutto, L. S. Dolci, A. Buragina, S. Prati, M. Guardigli, R. Mazzeo and A. Roda, Anal. Bioanal. Chem., 2011, 399, 2889–2897 Search PubMed.
- B. Ramírez-Barat and S.d. l. Viña, Stud. Conserv., 2001, 46, 282–288 Search PubMed.
- L. Kockaert, P. Gausset and M. Dubi-Rucquoy, Stud. Conserv., 1989, 34, 183–188 Search PubMed.
- P. L. Jones, Stud. Conserv., 1962, 7, 10–16 Search PubMed.
- M. Johnson and E. Packard, Stud. Conserv., 1971, 16, 145–164 Search PubMed.
- L. Cartechini, M. Vagnini, M. Palmieri, L. Pitzurra, T. Mello, J. Mazurek and G. Chiari, Acc. Chem. Res., 2010, 43, 867–876 Search PubMed.
- A. Nevin, S. Cather, D. Anglos and C. Fotakis, Anal. Chim. Acta, 2006, 573–574, 341–346 Search PubMed.
- M. Odlyha, Thermochim. Acta, 1995, 269–270, 705–727 Search PubMed.
- R. J. Meilunas, J. G. Bentsen and A. Steinberg, Stud. Conserv., 1990, 35, 33–51 CrossRef CAS.
- M. T. Doménech-Carbó, F. B. Reig, J. V. G. Adelantado and V. P. Martínez, Anal. Chim. Acta, 1996, 330, 207–215 CrossRef.
- A. Sarmiento, M. Perez-Alonso, M. Olivares, K. Castro, I. Martinez-Arkarazo, L. A. Fernandez and J. M. Madariaga, Anal. Bioanal. Chem., 2011, 399, 3601–3611 Search PubMed.
- E. Manzano, N. Navas, R. Checa-Moreno, L. Rodriguez-Simon and L. F. Capitan-Vallvey, Talanta, 2009, 77, 1724–1731 Search PubMed.
- A. Nevin, I. Osticioli, D. Anglos, A. Burnstock, S. Cather and E. Castellucci, Anal. Chem., 2007, 79, 6143–6151 CrossRef CAS.
- J. Peris-Vicente, E. Simó-Alfonso, J. V. Gimeno Adelantado and M. T. Doménech-Carbó, Rapid Commun. Mass Spectrom., 2005, 19, 3463–3467 Search PubMed.
- C. Tokarski, E. Martin, C. Rolando and C. Cren-Olivé, Anal. Chem., 2006, 78, 1494–1502 CrossRef CAS.
- C. Solazzo, W. W. Fitzhugh, C. Rolando and C. Tokarski, Anal. Chem., 2008, 80, 4590–4597 CrossRef CAS.
- W. Fremout, M. Dhaenens, S. Saverwyns, J. Sanyvova, P. Vandenabeele, D. Deforce and L. Moens, Anal. Chim. Acta, 2010, 658, 156–162 CrossRef CAS.
- G. Leo, L. Cartechini, P. Pucci, A. Sgamellotti, G. Marino and L. Birolo, Anal. Bioanal. Chem., 2009, 395, 2269–2280 CrossRef CAS.
- S. Kuckova, R. Hynek and M. Kodicek, J. Cult. Heritage, 2009, 10, 244–247 Search PubMed.
- A. Chambery, A. Di Maro, C. Sanges, V. Severino, M. Tarantino, A. Lamberti, A. Parente and P. Arcari, Anal. Bioanal. Chem., 2009, 395, 2281–2291 CrossRef CAS.
- W. Fremout, S. Kuckova, M. Crhova, J. Sanyova, S. Saverwyns, R. Hynek, M. Kodicek, P. Vandenabeele and L. Moens, Rapid Commun. Mass Spectrom., 2011, 25, 1631–1640 CrossRef CAS.
- C. D. Calvano, I. D. van der Werf, F. Palmisano and L. Sabbatini, Anal. Bioanal. Chem., 2011, 400, 2229–2240 Search PubMed.
- M. T. Doménech-Carbó, Anal. Chim. Acta, 2008, 621, 109–139 CrossRef CAS.
- J. de la Cruz-Cañizares, M. T. Doménech-Carbó, J. V. Gimeno-Adelantado, R. Mateo-Castro and F. Bosch-Reig, J. Chromatogr., A, 2004, 1025, 277–285 CrossRef CAS.
- M. P. Colombini, F. Modugno and A. Giacomelli, J. Chromatogr., A, 1999, 846, 101–111 CrossRef CAS.
- M. R. Schilling, H. P. Khanjian and L. A. C. Souza, J. Am. Inst. Conserv., 1996, 35, 45–59 Search PubMed.
- C. Marinach, M.-C. Papillon and C. Pepe, J. Cult. Heritage, 2004, 5, 231–240 CrossRef.
- E. Kouloumpi, G. Lawson and V. Pavlidis, Anal. Bioanal. Chem., 2007, 387, 803–812 CrossRef CAS.
- M. P. Colombini, F. Modugno, M. Giacomelli and S. Francesconi, J. Chromatogr., A, 1999, 846, 113–124 CrossRef CAS.
- R. M. Castro, M. T. Doménech-Carbó, V. P. Martínez, J. V. G. Adelantado and F. B. Reig, J. Chromatogr., A, 1997, 778, 373–381 CrossRef CAS.
- A. Casoli, P. C. Musini and G. Palla, J. Chromatogr., A, 1996, 731, 237–246 CrossRef CAS.
- I. Bonaduce, M. Cito and M. P. Colombini, J. Chromatogr., A, 2009, 1216, 5931–5939 CrossRef CAS.
- Z.-H. Huang, J. Wang, D. A. Gage, J. Throck Watson, C. C. Sweeley and P. Husek, J. Chromatogr., A, 1993, 635, 271–281 CrossRef.
- P. Bocchini and P. Traldi, J. Mass Spectrom., 1998, 33, 1053–1062 CrossRef CAS.
- G. Gautier and M. P. Colombini, Talanta, 2007, 73, 95–102 CrossRef CAS.
- A. Andreotti, I. Bonaduce, M. P. Colombini, F. Modugno and E. Ribechini, Int. J. Mass Spectrom., 2009, 284, 123–130 Search PubMed.
- E. Kenndler, K. Schmidtbeiwl, F. Mairinger and M. Pohm, Fresenius' J. Anal. Chem., 1992, 342, 135–141 CrossRef CAS.
- M. P. Colombini, R. Fuoco, A. Giacomelli and B. Muscatello, Stud. Conserv., 1998, 43, 33–41 Search PubMed.
- A. Andreotti, I. Bonaduce, M. P. Colombini, G. Gautier, F. Modugno and E. Ribechini, Anal. Chem., 2006, 78, 4490–4500 CrossRef CAS.
- G. Chiavari, D. Fabbri, G. C. Galletti, S. Prati and N. Scianna, J. Cult. Heritage, 2006, 7, 67–70 Search PubMed.
- M. Carbini, R. Stevanato, M. Rovea, P. Traldi and D. Favretto, Rapid Commun. Mass Spectrom., 1996, 10, 1240–1242 CrossRef CAS.
- G. Chiavari, G. C. Galletti, G. Lanterna and R. Mazzeo, J. Anal. Appl. Pyrolysis, 1993, 24, 227–242 Search PubMed.
- M. P. Colombini and F. Modugno, J. Sep. Sci., 2004, 27, 147–160 Search PubMed.
- M. P. Colombini, A. Andreotti, I. Bonaduce, F. Modugno and E. Ribechini, Acc. Chem. Res., 2010, 43, 715–727 Search PubMed.
- B. Carratù, C. Boniglia and G. Bellomonte, J. Chromatogr., A, 1995, 708, 203–208 CrossRef CAS.
- S. Hou, H. He, W. Zhang, H. Xie and X. Zhang, Talanta, 2009, 80, 440–447 CrossRef.
- P. Přikryl, L. Havlíčková, V. Pacáková, J. Hradilová, K. Štulík and P. Hofta, J. Sep. Sci., 2006, 29, 2653–2663 Search PubMed.
- S. M. Halpine, Stud. Conserv., 1992, 37, 22–38 Search PubMed.
- F. Ronca, Stud. Conserv., 1994, 39, 107–120 Search PubMed.
- J. Wouters, M. V. Bos and K. Lamens, Stud. Conserv., 2000, 45, 106–116 Search PubMed.
- K. Petritis, C. Elfakir and M. Dreux, J. Chromatogr., A, 2002, 961, 9–21 CAS.
- S. L. Vallance, Analyst, 1997, 122, 75R–81R RSC.
- D. Yan, G. Li, X.-H. Xiao, X.-P. Dong and Z.-L. Li, J. Chromatogr., A, 2007, 1138, 301–304 CrossRef CAS.
- M. Piraud, C. Vianey-Saban, K. Petritis, C. Elfakir, J. P. Steghens and D. Bouchu, Rapid Commun. Mass Spectrom., 2005, 19, 1587–1602 CrossRef CAS.
- P. Chaimbault, K. Petritis, C. Elfakir and M. Dreux, J. Chromatogr., A, 1999, 855, 191–202 CrossRef CAS.
- M. Piraud, C. Vianey-Saban, C. Bourdin, C. Acquaviva-Bourdain, S. Boyer, C. Elfakir and D. Bouchu, Rapid Commun. Mass Spectrom., 2005, 19, 3287–3297 CrossRef CAS.
- M. Armstrong, K. Jonscher and N. A. Reisdorph, Rapid Commun. Mass Spectrom., 2007, 21, 2717–2726 CrossRef CAS.
- K. Petritis, P. Chaimbault, C. Elfakir and M. Dreux, J. Chromatogr., A, 2000, 896, 253–263 CrossRef CAS.
- J. Qu, Y. Wang, G. Luo, Z. Wu and C. Yang, Anal. Chem., 2002, 74, 2034–2040 CrossRef CAS.
- M. Zoppa, L. Gallo, F. Zacchello and G. Giordano, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2006, 831, 267–273 CrossRef CAS.
- M. J. Desai and D. W. Armstrong, J. Mass Spectrom., 2004, 39, 177–187 CrossRef CAS.
- R. Kostiainen and T. J. Kauppila, J. Chromatogr., A, 2009, 1216, 685–699 Search PubMed.
- T. M. Annesley, Clin. Chem., 2003, 49, 1041–1044 CrossRef CAS.
- U. A. Mirza and B. T. Chait, Anal. Chem., 1994, 66, 2898–2904 CrossRef CAS.
- J.-Y. Kwon and M. Moini, J. Am. Soc. Mass Spectrom., 2001, 12, 117–122 CrossRef CAS.
- F. E. Kuhlmann, A. Apffel, S. M. Fischer, G. Goldberg and P. C. Goodley, J. Am. Soc. Mass Spectrom., 1995, 6, 1221–1225 CrossRef CAS.
- A. Apffel, S. Fischer, G. Goldberg, P. C. Goodley and F. E. Kuhlmann, J. Chromatogr., A, 1995, 712, 177–190 CrossRef CAS.
- R. Zangrando, R. Piazza, W. R. L. Cairns, F. C. Izzo, A. Vianello, E. Zendri and A. Gambaro, Anal. Chim. Acta, 2010, 675, 1–7 Search PubMed.
- M. Fountoulakis and H.-W. Lahm, J. Chromatogr., A, 1998, 826, 109–134 CrossRef CAS.
- M. Weiss, M. Manneberg, J.-F. Juranville, H.-W. Lahm and M. Fountoulakis, J. Chromatogr., A, 1998, 795, 263–275 CrossRef CAS.
- M. V. Pickering and P. Newton, Lc Gc-Magazine of Separation Science, 1990, 8, 778 Search PubMed.
- A. Jurado-Lopez and M. D. L. de Castro, Talanta, 2005, 65, 1059–1062 CrossRef CAS.
- P. Husek, J. Chromatogr., Biomed. Appl., 1998, 717, 57–91 CrossRef CAS.
- M. Delport, S. Maas, S. W. van der Merwe and J. B. Laurens, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2004, 804, 345–351 CrossRef CAS.
- J. J. Kamphorst, J. Fan, W. Lu, E. White and J. D. Rabinowitz, Anal. Chem., 2011, 83, 9114–9122 CrossRef CAS.
- K. Mitani, M. Fujioka, A. Uchida and H. Kataoka, J. Chromatogr., A, 2007, 1146, 61–66 CrossRef CAS.
- D. W. McMartin, K. M. Peru, J. V. Headley, M. Winkler and J. A. Gillies, J. Chromatogr., A, 2002, 952, 289–293 CrossRef CAS.
- T. Wyttenbach, M. Witt and M. T. Bowers, J. Am. Chem. Soc., 2000, 122, 3458–3464 CrossRef CAS.
- A. G. Harrison, Mass Spectrom. Rev., 1997, 16, 201–217 CrossRef CAS.
- M.a. H. Amad, N. B. Cech, G. S. Jackson and C. G. Enke, J. Mass Spectrom., 2000, 35, 784–789 CrossRef CAS.
- J.-P. Rauha, H. Vuorela and R. Kostiainen, J. Mass Spectrom., 2001, 36, 1269–1280 Search PubMed.
- G. Leo, I. Bonaduce, A. Andreotti, G. Marino, P. Pucci, M. P. Colombini and L. Birolo, Anal. Chem., 2011, 83, 2056–2064 Search PubMed.
- M. P. Colombini, F. Modugno, E. Menicagli, R. Fuoco and A. Giacomelli, Microchem. J., 2000, 67, 291–300 CrossRef CAS.
- O. F. van den Brink, J. J. Boon, P. B. O'Connor, M. C. Duursma and R. M. A. Heeren, J. Mass Spectrom., 2001, 36, 479–492 CrossRef CAS.
- O. F. van den Brink, E. S. B. Ferreira, J. van der Horst and J. J. Boon, Int. J. Mass Spectrom., 2009, 284, 12–21 Search PubMed.
Footnotes |
| † Presented at the VIIIth Polish conference of Analytical Chemistry. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ay05605d |
| This journal is © The Royal Society of Chemistry 2012 |