EndoV/DNA ligase mutation scanning assay using microchip capillary electrophoresis and dual-color laser-induced fluorescence detection
Akira
Kotani
*ab,
Małgorzata A.
Witek
b,
John K.
Osiri
b,
Hong
Wang
b,
Rondedrick
Sinville
b,
Hanna
Pincas
c,
Francis
Barany
c and
Steven A.
Soper
bdef
aSchool of Pharmacy, Tokyo University of Pharmacy and Life Sciences, 1432-1 Horinouchi, Hachioji, Tokyo, 192-0392, Japan. E-mail: kotani@toyaku.ac.jp; Fax: +81-42-676-4570; Tel: +81-42-676-4569
bDepartment of Chemistry, Louisiana State University, Baton Rouge, LA 70803, USA
cDepartment of Microbiology and Immunology, Weill Cornell Medical College, New York, NY 10065, USA
dDepartment of Mechanical Engineering, Louisiana State University, Baton Rouge, LA 70803, USA
eThe Center for Biomodular Multi-Scale Systems, Louisiana State University, Baton Rouge, LA 70803, USA
fUlsan National Institute of Science and Technology, Ulsan Metropolitan City, 689-798, Republic of Korea
First published on 7th October 2011
Abstract
We report the ability to detect with high sensitivity sporadic mutations using a mutation scanning assay, which employs thermostable endonuclease V (EndoV) and DNA ligase. The products of the mutation scanning assay were separated using microchip capillary electrophoresis (μCE) and detected with a dual-color laser-induced fluorescence (LIF) detector. PCR products from mutant and wild-type DNA of p53 exon 8 were generated using Cy3-labeled forward and Cy5-labeled reverse primers to allow LIF detection with μCE. EndoV recognizes and primarily cleaves heteroduplexed DNA one base 3′ to a mismatch and can nick matched sites at low levels as well. DNA ligase is used to reseal nicks generated at matched sites, which creates a highly sensitive and specific assay for analyzing sporadic mutations in genomic DNA. Heteroduplexed DNA samples were treated with EndoV alone and with both EndoV and DNA ligase and separated using a 4% (w/v) linear polyacrylamide gel constituted in 1x TTE buffer, 7 M urea, and 0.05% (w/v) methyl hydroxyethyl cellulose, which was used to suppress the EOF in the microchip. Sizing of the bands appearing in the electropherogram revealed the approximate position of the mutation. In this study, mutations present in p53 exon 8 generated Cy3-labeled cleavage products of 158 nt and Cy5-labeled cleavage products of 195 nt. The DNA fragments were simultaneously monitored at their respective color using a dual-color LIF system with the 158 and 195 nt fragments detected along with heteroduplexed fragments of 350 nt. The microchip separation was completed within 7 min, almost ten-fold shorter time compared to conventional capillary gel electrophoresis.
Introduction
Accumulation of mutations stemming either from inherited or somatic alterations are ultimately responsible for spawning most cancer-related diseases. Overall, approximately 90% of all cancers are attributed to somatic mutations, ∼20% are the result of germline mutations and ∼10% involve both types.1 The Human Genome Project (HGP) resulted in the identification and characterization of many genetic alterations associated with cancer.2 This information has provided insights into the structure and function of oncogenes and tumor suppressor genes, both of which play a pivotal role in tumorigenesis. Oncogenes produce growth and signal transduction factors that promote cell growth and proliferation,3 while active tumor suppressor genes, which are often found deactivated in many cancer-related diseases, regulate cell growth and initiate apoptosis in cells.4,5Most mutations associated with a particular patient's cancer must be accurately mapped and evaluated in clinical settings to realize viable diagnostic and prognostic information that can guide therapy as well as determine response to therapy and monitor recurrence. For example, KRAS mutations in codons 12 and 13 occur in 80–90% of pancreatic cancers and 35–50% of colorectal cancers;6–8single nucleotide polymorphisms in BRCA1 and BRCA2 present at low frequencies (1–5%) are linked to significantly higher risks of developing breast, ovarian or prostate cancers for certain ethnic groups.9 A locus in chromosome region 15q25 encompassing several genes - including three that encode nicotinic acetylcholine receptor subunits (CHRNA3, CHRNA4, and CHRNA5) - have been found to account for 14% of lung cancer cases.10 The location of these mutations are known and thus, can be analyzed using mutation detection assays, such as allele specific ligation,11allele specific PCR12 or allele-specific hybridization.13
The presence or likelihood of developing cancer-related diseases have also been associated with the presence of sporadic p53 (tumor suppressor gene) mutations,14 which are mutations that can occur any place within a certain gene and are not confined to a particular locus, making them difficult to detect.15 A further challenge in evaluating sporadic mutations is that they are often inundated with excess wild-type DNA (wtDNA) in clinical samples of early stage cancer diseases as well as the number of mutations can be highly variable. For example, the frequency of sporadic p53 mutations in exons 5–9 can be as low as one mutant DNA (mtDNA) per 1,000 wild-type sequences.16 In addtion, over 22,000 p53 mutations in different human cancers have been recorded and compiled, which can be found in an accessible database.17
Several methods have been developed to detect sporadic mutations including hybridization analysis using high-density oligonucleotide arrays,18 denaturing high-performance liquid chromatography (DHPLC),19,20 single strand conformational polymorphism (SSCP),21denaturing gradient gel electrophoresis (DGGE),22 heteroduplex analysis (HA),23 and dideoxy-sequencing.24 Unfortunately, none of these methods have the combined ability to provide low detection limits when wtDNA is in a large excess and the exact location of the sporadic mutation(s) is not provivded by the assay. Although Sanger dideoxy-sequencing can detect any single nucleotide polymorphism and determine its location, the procedure is often time consuming and lacks the necessary sensitivity to detect low abundance mtDNA in large excesses of wtDNA.
An approach for determining the approximate location of sporadic mutations even in the presence of large excesses of wtDNA is the use of enzymatic approaches, such as T4 Endonuclease VII, which can cleave mutational sites within double-stranded (ds) DNA.25,26 However, this method generally suffers from miscleavages leading to false positive signals.
Recently, a one-step mutation scanning assay has been reported (Fig. 1), which employs Thermotoga maritimaendonuclease V (EndoV).27 EndoV is an enzyme that clips a dsDNA molecule containing mismatched base pairs (i.e., heteroduplexes) on the 3′ side of the mismatch. Unfortunately, EndoV can also nick dsDNA at matched sites, which can generate false positive signals when attempting to transduce the presence and location of mismatches. However, one can repair the nicks at matched sites using Thermus AK16D thermostable DNA ligase, which offers 5-fold better discriminatory power for matched sites compared to other ligases,28–30 thereby reducing background signals generated from matched sites cleaved by EndoV.27
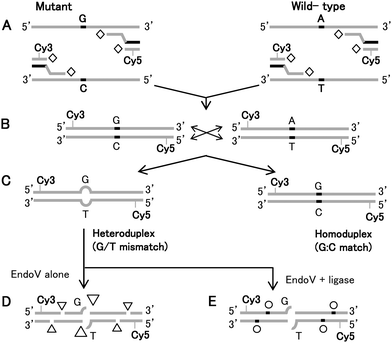 | ||
| Fig. 1 (A) Schematic illustration of universal PCR amplification using Taq DNA polymerase (open diamond), labeling and (B) cross-pairing of wild-type Lovo and mutagenic SW620 template and (C) the resulting homoduplex (control) and heteroduplex targets. The diagram illustrates the outcome of a successful EndoV/DNA ligase treatment of dye-labeled, heteroduplexed DNAs. PCR amplicons of known wild-type and mutagenic cell lines having high levels of sporadic mutations, form heteroduplexes serving as targets for EndoV. (D) EndoV preferentially nicks DNA one base 3′ to the mismatch site (large triangle), but also generates non-specific nicks with minor activity (small triangles). (E) DNA ligase is then used either subsequently or concurrently with EndoV to reseal background nicks (open circle). | ||
By combining EndoV with AK16D thermostable DNA ligase,31,32 an EndoV/DNA ligase assay has been shown to provide a sensitivity of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 (mtDNA:wtDNA) for scanning for the presence of sporadic mutations in any gene. Furthermore, this strategy employs a universal PCR amplification step making it amenable to multiplexing.27 The terminal step required in this EndoV/DNA ligase assay is high-resolution electrophoresis that sizes the DNAs in their single-stranded form to determine the location of the sporadic mutation site.
:50 (mtDNA:wtDNA) for scanning for the presence of sporadic mutations in any gene. Furthermore, this strategy employs a universal PCR amplification step making it amenable to multiplexing.27 The terminal step required in this EndoV/DNA ligase assay is high-resolution electrophoresis that sizes the DNAs in their single-stranded form to determine the location of the sporadic mutation site.
The trend towards miniaturizing electrophoretic platforms for DNA separations was initiated during the HGP and has impacted genetic analyses for clinical diagnostics/prognostics by providing high-resolution separations in short electrophoretic development times. This has led to the development of microchip capillary electrophoresis (μCE), which possesses simple operational characteristics and can be seamlessly integrated to frontend sample processing strategies. Efforts have prompted the engineering of various high-throughput and highly integrated electrophoretic devices in glass. Recently, polymeric substrates, which are inexpensive materials conducive to a variety of high production-mode fabrication techniques, have been developed producing devices appropriate for clinical applications. As opposed ot research applications, clinical usage requires one-time use devices to prevent false positive signals arising from sample carryover contamination.33–36
Mutation scanning assays, which usually depend on an electrophoretic separation for reading results from the molecular assay, have been demonstrated using μCE. For example, mutation scanning assays that have been transitioned to microchip platforms include SSCP and HA.37–39 Many of these reports emphasized numerous operational parameters, such as electrophoresis temperature and polymer matrix/denaturing additive concentrations that must be optimized to provide favorable results for mutational analyses possessing adequate resolution for detecting the target mutations.40–42 However, the use of the EndoV/DNA ligase mutation scanning assay has not been demonstrated using μCE to-date in spite of its appealing operational characteristics compared to capillary or conventional slab gel electrophoresis used for the EndoV/Ligase assay.27
The EndoV/DNA ligase mutation scanning assay requires dual-color laser-induced fluorescence (LIF) detection because two different dye-labeled ssDNA fragments must be simultaneously detected; both the upper and lower strands must be analyzed to ascertain whether the mutation(s) were from either the upper or lower strand following EndoV/DNA ligase processing. Recently, a dual-color μCE system with single-photon avalanche diodes (SPADs) was reported, which was applied for the detection of a mutation associated with cystic fibrosis; ΔF508.43,44
Herein, we describe the highly efficient separation and sensitive detection of EndoV/DNA ligase reaction products for mutation scanning using μCE with dual-color LIF detection. The microchip for the electrophoresis was made from poly(methylmethacrylate), PMMA. We will demonstrate the utility of this assay and system for identifying sporadic p53 mutations in a model system, which consisted of immortalized cells lines with a known KRAS mutational status.
Experimental
Materials
Genomic DNA was extracted from Lovo and SW620 cell lines having known KRAS oncogenic mutations using a genomic DNA miniprep kit (GenElute Mammalian, Sigma-Aldrich, St. Louis, MO, USA); the Lovo cell line contains a wt p53 gene and the SW620 cell line contains an exon 8 R273H (G → A) mutation.PCR amplification
The sequences of the PCR primers used in this study are listed in Table 1. We used a universal labeling strategy with the dyes locallized internally on the primer sequence,27 which is amenable to multiplexed amplification of dozens of genes, addressing the practical reality that tumor DNA is often limiting. This strategy employs two sets of primer pairs; unlabeled gene-specific primers that harbor universal tails on their 5′ ends (see Table 1 and Fig. 1) and fluorescently-labeled universal primers that can be used for any gene/exon amplification (Fig. 1A). PCRs for amplification of specific genes were carried out using universal primer amplification cocttails (50 μL) containing 15 mM Tris/HCl (pH 8.05), 50 mM KCl, 2.5 mM MgCl2, 0.2 mM of dNTPs, 0.2 μM of each universal primer, 0.02 μM of gene-specific primers for p53 exon 8, 1.25 U of Ampli Taq Gold DNA polymerase and 150 ng of genomic DNA. Thermal cycling conditions were as follows: 95 °C for 10 min to activate Ampli Taq Gold polymerase, followed by 20 cycles of 94 °C for 30 s, 65 °C for 1 min, 72 °C for 1 min (for gene-specific amplification) and then 30 cycles of 94 °C for 30 s, 55 °C for 1 min, 72 °C for 1 min (for universal amplification), followed by a final extension step at 72 °C for 7 min. During the first 20 PCR cycles, an annealing temperature of 65 °C allowed gene specific primers at low concentration to anneal selectively to their target DNA template, while the universal primers hybridize to gene-specific primer universal tails. By shifting the annealing temperature to 55 °C for an additional 30 PCR cycles, the Cy3- and Cy5-labeled universal primers predominate in amplifying labeled products. For the universal PCR, the universal primer pair consisted of a forward Cy3-labeled primer and a reverse Cy5-labeled primer.| Gene | Primer name | Primer sequence |
|---|---|---|
| a F, forward primer; R, reverse primer. Bases highlighted in bold correspond to the universal sequence. | ||
| Universal | Cy3_UniEV1F | 5′ CGC C (c7-NH- Cy3) GT CAC GAC ACG AAA AC 3′ |
| Universal | Cy5_UniEV2R | 5′ CGC C (c7-NH- Cy5) GT CAC GAC ACG AAA CA 3 |
| p53 exon 8 | F173 | 5′ CGT CAC GAC ACG AAA ACC AGG GTG GTT GGG AGT AGA TG 3′ |
| p53 exon 8 | R174 | 5′ CGT CAC GAC ACG AAA CAG GTG ATA AAA GTG AAT CTG AGG CAT AAC 3′ |
Preparation of heteroduplexed DNA substrates
For heteroduplex generation, approximately equal ratios of Cy3/Cy5-labeled wild-type PCR amplicons (Lovo) were mixed with Cy3/Cy5-labeled mutant PCR amplicons (SW620) to a 12 μL final volume (150 ng total DNA). The homoduplex (wild-type control) consisted of Cy3/Cy5-labeled wild-type PCR products alone in a 12 μL final volume. The residual Taq DNA polymerase was inactivated by adding 1 μL of proteinase K (20 μg mL−1) to each mixture and incubating at 65 °C for 30 min, followed by a 10 min incubation at 80 °C to inactivate proteinase K. For denaturation/renaturation, PCR mixtures were heated at 95 °C for 2 min and gradually cooled to room temperature with a 0.2 °C decrease in temperature every 15 s to 45 °C, and finally a 10 min incubation at 25 °C to generate heteroduplexes (Fig. 1C).EndoV/DNA ligase mutation scanning assay
A 6.5–μL volume of each denatured/renatured PCR mixture was incubated at 65 °C for 12 h in a 20 μL volume reaction cocktail containing 80 mM tricine (pH 8.0), 5 mM MgCl2, 5 mM dithiothreitol (DTT), 1.5 M betain, 2% glycerol with 0.5 U/μL EndoV (EN01401, Fermentans Inc., Glen Burnie, MD, USA), 1.6 U/μL Taq DNA ligase (M0208L, New England BioLabs, Ipswich, MA, USA) and 1 mM nicotinamide adenine dinucleotide (NAD). The reactions were terminated with the addition of EDTA to a 10 mM final concentration to sequester EndoV cleavage activity. Using these conditions, both EndoV cleavage and ligase DNA repair of miscleavage events were carried out simultaneously.Microchip fabrications
PMMA was selected as the μCE substrate material because of its suitable physicochemical properties appropriate for this application, such as minimal non-specific adsorption artifacts and low levels of autofluorescence, improving the limit-of-detection to allow inspection of low copy number input samples.45 Microchips were made using procedures described previously.33 Briefly, microstructures were micromilled into a brass plate 0.25′′ thick alloy 353 engraver brass, McMaster-Carr, Atlanta, GA, USA) using a Kern MMP 2522 micromilling machine (KERN Mikro-und Feinwerktechnik GmbH, Germany). Once fabricated, the mold master produced PMMA replicas formed via hot embossing. Hot embossing required heating the molding tool to 160 °C, which was then pressed into the PMMA plate using a pressure of 1,100 psi for 410 s (PHI Precision Press, PHI-Tulip, City of Industry, CA, USA). Following embossing, the PMMA substrate was cooled to room temperature and removed from the molding tool. The embossed PMMA substrate was cleaned with 50% isopropanol in ultrapure water. Finally, a PMMA cover plate 0.125 mm) was thermally fusion bonded to the substrate by heating in a temperature programmable furnace to 107 °C, slightly above the glass transition temperature (Tg) of PMMA. The microstructures produced a separation channel with dimensions of 70 μm (depth) × 30 μm (width) that was 9.5 cm long with 0.5 cm intersecting side channels offset by 0.5 mm (Fig. 2), which provided a defined ∼1 nL volume for sample injection. | ||
| Fig. 2 Schematic of the PMMA microchip. The chip contained four reservoirs, A – sample; B – waste; C – buffer, and D – buffer waste. The chip also contained a separation channel that was 10 cm in total length with a channel width of 30 μm and depth of 70 μm. The effective channel separation length was 3.5 cm. | ||
Dual-color LIF instrumentation
The dual-color LIF detection system was built with an epi-illumination configuration as shown in Fig. 3. It was desinged to monitor the emmisions of Cy3 (excitation wavelength (ex)/emission wavelength (em) 550 nm/570 nm) and Cy5 (ex/em 649 nm/670 nm) for each respective color channel. The excitation source consisted of a 532 nm diode laser (20 mW, SLL-532-DT020-LM, OEM Laser System, East Lansing, MI, USA) and a 635 nm diode laser (3 mW, 56-DIB-142/P1, CVI Melles Griot, Albuquerque, NM, USA). The 532 nm and 635 nm lasers were filtered by a laser line filter (LLF1, CWL = 532 nm, XL08, Omega Optical, Brattleboro, VT, USA; and LLF2, CWL = 635 nm, XL37, Omega Optical). A dichroic mirror (DM1, XF2018, Omega Optical) reflected the 532 nm and transmitted the 635 nm lasers wavelengths. Both laser beams were directed into a focusing objective using a second dichroic mirror (DM2; XF2055, Omega Optical). The excitation beams were focused using a microscope objective (Nikon, Natick, MA, USA) into the separation channel of the microchip, which was situated on an X-Y-Z microtranslational stage (Newport, Irvine, CA, USA).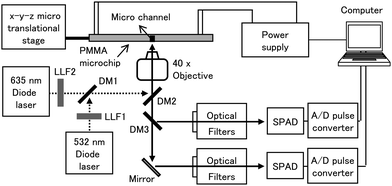 | ||
| Fig. 3 Schematic diagram of the LIF system, which provided two excitation wavelengths of 532 and 635 nm. LLF1 and 2, laser line filters; DM1, 2, and 3; dichronic mirrors; SPAD, single photon avalanche diode. | ||
The resulting emission from Cy3-labeled products was collected by the microscope objective and routed through a dichroic mirror (DM2), reflected onto another dichroic mirror (DM3, DMLP605, Thorlabs, Newton, NJ, USA), and finally filtered through a long pass filter (CWL = 550 nm, 3RD550LP, Omega Optical) and a band pass filter (CWL = 570 nm, XB99, Omega Optical) with the resulting photons transduced using a single photon avalanche diode (SPAD; SPCM 200B, PicoQuant, Berlin, Germany). The resulting emission from Cy5 was routed through DM2 and DM3, then reflected by a mirror and filtered through a long pass filter (CWL = 650 nm, 3RD650LP, Omega Optical) and a band pass filter (CWL = 670 nm, XB114, Omega Optical) before being processed by a second SPAD. The LIF signals were acquired on a personal computer equipped with an I/O interface board (CB-68LP, National Instruments, Austin, TX, USA) and a pulse converter (TB-01, IBH, Glasgow, UK). Data acquisition software was written in LabView.
A custom Labview program was used to control applied voltages for the microchip electrophoresis. This unit included three internal high voltage power supplies (EMCO, Sutter Creek, CA) capable of receiving inputs of 0 or +5 V from a DAC (digital-to-analog converter) output from a CB-68LP board (National Instruments). These power supplies were capable of delivering 0 to +2 kV to sample and waste reservoirs (EMCO Model C20, Sutter Creek, CA) and +0.3 to +5 kV to the anodic reservoir (EMCO Model G50), all of which could be dynamically altered throughout the separation.
Capillary and microchip capillay electrophoresis conditions for EndoV/DNA ligase assay
Separation of the EndoV/DNA ligase products using conventional capillary gel electrophoresis was performed with a CEQ8000 system obtained from Beckman Coulter (Fullerton, CA, USA) equipped with a 4-channel detector (ex/em 1–650 nm/670 nm; 2–650 nm/706 nm; 3–750 nm/770 nm; 4–750 nm/785 nm) and eight capillaries (id = 75 μm) with an effective separation length of 30 cm. Samples were diluted 1:40 with HiDi formamide and 0.25 μL of a CEQ DNA size standard 400 (Beckman Coulter), which were denaturated at 95 °C for 2 min to ensure all duplexes were converted into the single stranded form and then, electrokinetically loaded into the capillary by applying 2.0 kV for 30 s with electrophoresis carried out at 7.5 kV. During the electrophoresis, the temperature of the capillary was maintained at 60 °C.
The μCE separations were performed at room temperature using microchannels filled with a 4% (w/v) linear polyacrylamide (LPA) suspended in 1X TTE buffer (50 mM Tris, 50 mM TAPS, 2 mM EDTA) and 7 M urea containing 0.05% (w/v) methyl hydroxyethyl cellulose (MHEC, Sigma Aldrich, St. Louis, MO). The MHEC served to dynamically coat the PMMA channel walls for electroosmotic flow (EOF) suppression.46 One μL of the reaction cocktail was mixed with 1 μL HiDi formamide and introduced on chip. To generate a volume-defined injection plug, +550 V was applied for 60 s from the sample reservoir (ground) to the waste reservoir (Fig. 2). Electrophoresis of the sample was conducted using a 125 V cm−1 electric field strength, while pull back field strengths of 90 V cm−1 were applied to the sample and waste reservoirs to prevent extraneous sample from leaking into the separation channel during the electrophoresis.
Results and discussions
For these initial experiments, we utilized two cell lines, Lovo containing a wt p53 gene and the SW620 cell line, which contains an exon 8 R273H (G → A) mutation. While the location of the mutation in the SW620 cell line is known and could be analyzed via a mutation detection strategy, we used this cell line as a model for the detection of sporadic mutations requiring a mutation scanning assay because in clinical samples, the locations of p53 mutations are not known.Performance of LIF dual-color fluorescence detector
To evaluate possible cross-talk between detection channels, Cy3- or Cy5-labeled primers were injected into the μCE system. For injection, 350 V cm−1 was applied for 30 s from the sample reservoir (ground) to the waste reservoir (+350 V) to generate a volume-defined injection plug. When the Cy3-labeled forward primer was injected, a peak was only detected in the channel designed for Cy3 and no signal was observed in the channel designed for the detection of Cy5-labeled products. We applied the same procedure to Cy5-labeled primers and observed no cross talk in the Cy3 color channel. We estimated that the limit-of-detection for the Cy3- and Cy5-labeled primers (S/N = 3) were 12.6 and 13.4 pM, respectively.Universal PCR products
In order to ascertain whether the mutation(s) were present in the upper or lower strand, labeled primers with different fluorescent dyes were targeted for the upper and lower strands (see Fig. 1). This universal labeling strategy was implemented to eliminate the need for obtaining fluorescently-labeled primers for every loci that was to be interrogated. In this study, forward and reverse primers were internally labeled using Cy3 and Cy5, respectively, to amplify the gene-specific PCR products. Attachment of the fluorescent label internally in the primer sequencevia a C6 spacer rendered the products resistant to EndoV cleavage.27 To verify successful universal PCR amplification, amplicons were separated using a 2% agarose gel and compared to a size standard. The generation of 350 bp universal PCR products of wild-type and mutagenic targets were observed (data not shown).Capillary separation of EndoV/DNA ligase products
Prior to μCE analysis, separations of EndoV/DNA ligase products were conducted using a commercial CEQ8000 capillary gel electrophoresis system to ensure that the reactions were successful and to provide a comparison for the μCE results. If mutations were present in p53 exon 8, a Cy3-labeled cleavage product of 158 nt and Cy5-labeled cleavage product of 195 nt would be obtained from the heteroduplexed sample treated with both EndoV and DNA ligase, which have been confirmed viaDNA sequencing.47The EndoV treated samples in Fig. 4A showed elevated levels of sample cleavage resulting in several peaks that were <110 nt in size, which consisted of a mixture of primers and miscleaved products. However, when the universal PCR products were treated with both EndoV and DNA ligase (Fig. 4B), a reduction in the numer of false positive peaks generated from EndoV cleavage at fully matched sites was apparent. In fact, the peak intensities generated by EndoV miscleavages ranging between 100 and 190 nt (Fig. 4B) were significantly decreased compare to those seen in Fig. 4A. In the electropherogram of the heteroduplexed sample, which were treated with EndoV alone, a cleavage product peak was observed at 195 nt (Fig. 4A). Moreover, this peak intensity increased significantly after reaction with both EndoV and DNA ligase (Fig. 4B). Also, no product peak at 195 nt was present in wt control samples, as expected. From these results, it was concluded that cleavage products generated by EndoV/DNA ligase were obtained under the particular reaction conditions employed in these studies.
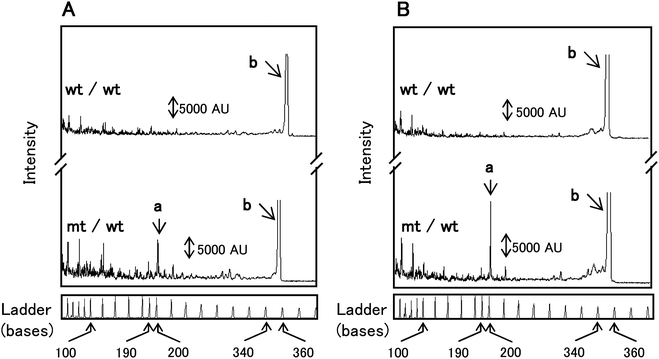 | ||
| Fig. 4 Capillary electrophoresis separation of wt control homoduplex (wt/wt) and heteroduplex (mt/wt) treated with (A) EndoV alone and (B) both EndoV and DNA ligase Peaks: a – Cy5 labeled cleavage products of 195 nt; b – PCR amplicon of 350 nt. Electrokinetic injections and separations were performed at 2.0 kV for 30 s and an applied voltage of 7.5 kV. | ||
In subsequent experiments starting with the universal PCR, Cy5-labeled cleavage products of 195 nt were also observed from all samples of mt/wt heteroduplexes were treated with EndoV alone. In additon, this 195 nt peak was observed when treated with EndoV and DNA ligase, but no product peak at 195 nt was observed in the wt control samples.
Microchip separation of EndoV/DNA ligase products
The μCE separation of wt control (homoduplexes) and a mt/wt heteroduplex sample treated with EndoV alone are shown in Fig. 5A. Most of the miscleavages resulted in several partially resolved fragments ranging in size from that of the excess primer used for the universal amplicon generation (∼20 nt) to approximately 110 nt. These peaks represent false positive signals due to non-specific cleavage of matched sites. The wt control (homoduplex) and the mt/wt (heteroduplex) dye-labeled amplicon products at 350 nt were observed at 375 s in each of the appropriate color channels.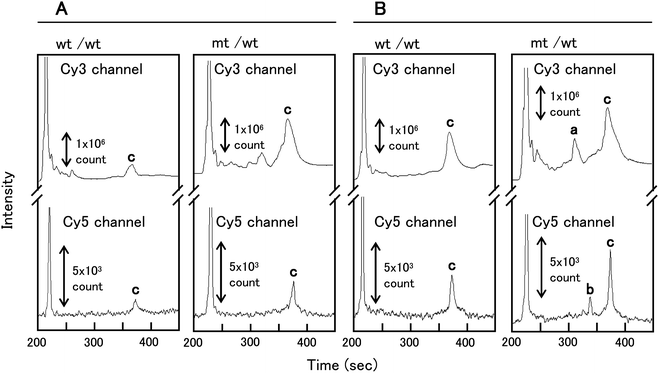 | ||
| Fig. 5 PMMA μCE separations of control homoduplexes (wt/wt) and heteroduplexes (mt/wt) treated with: (A) EndoV alone; and (B) both EndoV and DNA ligase. Peaks in the electropherogram are: a – Cy3-labeled cleavage products of 158 nt; b – Cy5-labeled cleavage products of 195 nt; c – PCR amplicon of 350 nt. The volume-defined cross injection (1 nL) and separation were performed at 550 V cm−1 (60 s) and 125 V cm−1, respectively. | ||
The μCE separation of wt control (homoduplex) and mt/wt heteroduplex samples treated concurrently with both EndoV and the DNA ligase are shown in Fig. 5B. Peak intensities for both the 350 nt wt/wt homoduplex and the mt/wt heteroduplex increased in intensity after reaction with both EndoV and DNA ligase as compared to the reaction with EndoV only. In the electropherogram of mt/wt heteroduplex samples treated with both EndoV and DNA ligase, a Cy3-labeled cleavage product at 158 nt and Cy5-labeled cleavage product at 195 nt were observed at 315 and 341 s, respectively, in the appropriate color channel (Fig. 5B). The reproducibily for the μCE separation of these clevage products were examined. The relative standard deviation (RSD, n = 3) of migration times for the Cy3- and Cy5-labeled cleavage products were 0.66% and 0.58%, respectively, indicating high separation reproducibility for the EndoV/DNA ligase products when using μCE. The μCE separation efficiency for the Cy5-labeled cleavage product of 195 nt was 7.4 × 105 plates/m, while the plate numbers for this same fragment was 8.2 × 105 plates/m for capillary gel electrophoresis. The efficiencies of the EndoV/DNA ligase product separations by μCE were similar in comparison to that obtained by capillary gel electrophoresis. The microchip separation was completed within a 7 min development time compared to approximately 1 h required for the separation of these products using conventional capillary gel electrophoresis.
Conclusions
We demonstrated the implementation of a mutation scanning assay that utilized EndoV to cleave duplexed DNA at the 3′ side of mismatched base pairs and also, indiscriminately nicked duplexed DNA at matched sites as well. However, the addition of a DNA ligase can repair EndoV nicks at matched sites, significantly improving the ability to detect sporadic mutations with high sensitivity, even in the presence of large amounts of wtDNA sequences, which existing mutation scanning assays such as SSCP cannot. To detect the location of the sporadic mutation, μCE was employed with dual-color LIF to determine if the mutation occurred on the top or bottom strand of the target DNA. Using the present system, Cy3- and Cy5-labeled ssDNA fragments were simultaneously detected within 7 min using μCE. Using a commercial capillary electrophoresis system, the same separation of Cy5-labeled ssDNA fragments required approximately 1 h. The present mutation scanning system along with dual-color LIF and μCE can achieve high-throughput detection of somatic mutations in tumors or the location of sporadic and/or unknown mutations in target genes, even when the amount of wtDNA sequences are high in the sample.Acknowledgements
The authors would like to thank the National Institutes of Health (National Cancer Institute, R21–1CA128671) for the financial support of this work.Notes and references
- P. A. Futreal, L. Coin, M. Marshall, T. Down, T. Hubbard, R. Wooster, N. Rahman and M. R. Stratton, Nat. Rev. Cancer, 2004, 4, 177 CrossRef CAS.
- R. Sachidanandam, D. Weissman, S. C. Schmidt, J. M. Kakoi, L. D. Stein, G. Marth and S. Sherry, et al. , Nature, 2001, 409, 928 CrossRef CAS.
- L. C. Cantley, K. R. Auger, C. Carpenter, B. Duckworth, A. Graziani, R. Kapeller and S. Soltoff, Cell, 1991, 64, 281 CrossRef CAS.
- S. Hietanen, S. Lain, E. Krausz, C. Blattner and D. P. Lane, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 8501 CrossRef CAS.
- R. A. Weinberg, Science, 1991, 254, 1138 CAS.
- K. Motojima, T. Urano, Y. Nagata, H. Shiku, T. Tsunoda and T. Kanematsu, Am. J. Gastroenterol., 1991, 86, 1784 CAS.
- C. P. Dieterle, M. Conzelmann, U. Linnemann and M. R. Berger, Clin. Cancer Res., 2004, 10, 641 CrossRef CAS.
- P. Anker, F. Lefort, V. Vasioukhin, J. Lyautey, C. Lederrey, X. Q. Chen, M. Stroun, H. E. Mulcahy and M. J. G. Farthing, Gastroenterology, 1997, 112, 1114 CrossRef CAS.
- J. P. Struewing, P. Hartge, S. Wacholder, S. M. Baker, M. Berlin, M. McAdams, M. M. Timmerman, L. C. Brody and M. A. Tucker, N. Engl. J. Med., 1997, 336, 1401 CrossRef CAS.
- R. J. Hung, J. D. McKay, V. Gaborieau, P. Boffetta, M. Hashibe, D. Zaridze and A. Mukeria, et al. , Nature, 2008, 452, 633 CrossRef CAS.
- D. Y. Wu, L. Ugozzoli, B. K. Pal and R. B. Wallace, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 2757 CrossRef CAS.
- A. Russom, A. Ahmadian, H. Andersson, P. Nilsson and G. Stemme, Electrophoresis, 2003, 24, 158 CrossRef CAS.
- B. J. Conner, A. A. Reyes, C. Morin, K. Itakura, R. L. Teplitz and R. B. Wallace, Proc. Natl. Acad. Sci. U. S. A., 1983, 80, 278 CrossRef CAS.
- K. A. Phillips, K. Nichol, H. Ozcelik, J. Knight, S. J. Done, P. J. Goodwin and I. L. Andrulis, J. Natl. Cancer Inst., 1999, 91, 469 CrossRef CAS.
- T. Soussi, K. Dehouche and C. Beroud, Hum. Mutat., 2000, 15, 105 CrossRef CAS.
- P. O. Ekstrom, A. L. Borresen-Dale, H. Qvist, K. E. Giercksky and W. G. Thilly, BioTechniques, 1999, 27, 128 CAS.
- D. Hamroun, S. Kato, C. Ishioka, M. Claustres, C. Beroud and T. Soussi, Hum. Mutat., 2006, 27, 14 CrossRef CAS.
- J. G. Hacia, J. B. Fan, O. Ryder, L. Jin, K. Edgemon, G. Ghandour, R. A. Mayer, B. Sun, L. Hsie, C. M. Robbins, L. C. Brody, D. Wang, E. S. Lander, R. Lipshutz, S. P. Fodor and F. S. Collins, Nat. Genet., 1999, 22, 164 CrossRef CAS.
- E. Schaeffeler, T. Lang, U. M. Zanger, M. Eichelbaum and M. Schwab, Clin. Chem., 2001, 47, 548 CAS.
- W. G. Liu, D. I. Smith, K. J. Rechtzigel, S. N. Thibodeau and C. D. James, Nucleic Acids Res., 1998, 26, 1396 CrossRef CAS.
- D. Glavac and M. Dean, Hum. Mutat., 1993, 2, 404 CrossRef CAS.
- R. Fodde and M. Losekoot, Hum. Mutat., 1994, 3, 83 CrossRef CAS.
- A. J. Nataraj, I. Olivos-Glander, N. Kusukawa and W. E. Highsmith, Electrophoresis, 1999, 20, 1177 CrossRef CAS.
- F. Sanger, S. Nicklen and A. R. Coulson, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 5463 CrossRef CAS.
- I. I. Haris, P. M. Green, D. R. Bentley and F. Giannelli, PCR Methods Appl., 1994, 3, 268 CAS.
- R. Youil, B. W. Kemper and R. G. H. Cotton, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 87 CrossRef CAS.
- H. Pincas, M. R. Pingle, J. Huang, K. Q. Lao, P. B. Paty, A. M. Friedman and F. Barany, Nucleic Acids Res., 2004, 32, e148 CrossRef.
- J. Tong, W. G. Cao and F. Barany, Nucleic Acids Res., 1999, 27, 788 CrossRef CAS.
- J. Huang, B. Kirk, R. Favis, T. Soussi, P. Paty, W. Cao and F. Barany, Oncogene, 2002, 21, 1909 CrossRef CAS.
- J. M. Huang, J. Lu, F. Barany and W. G. Cao, Biochemistry, 2002, 41, 8342 CrossRef CAS.
- M. Khanna, W. G. Cao, M. Zirvi, P. Paty and F. Barany, Clin. Biochem., 1999, 32, 287 CrossRef CAS.
- F. Barany, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 189 CrossRef CAS.
- M. L. Hupert, W. J. Guy, S. D. Llopis, H. Shadpour, S. Rani, D. E. Nikitopoulos and S. A. Soper, Microfluid. Nanofluid., 2007, 3, 1 CrossRef CAS.
- J. H. Aborn, S. A. El-Difrawy, M. Novotny, E. A. Gismondi, R. Lam, P. Matsudaira, B. K. Mckenna, T. O'Neil, P. Streechon and D. J. Ehrlich, Lab Chip, 2005, 5, 669 RSC.
- R. G. Blazej, P. Kumaresan and R. A. Mathies, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 7240 CrossRef CAS.
- R. Sinville and S. A. Soper, J. Sep. Sci., 2007, 30, 1714 CrossRef CAS.
- H. J. Tian, A. Jaquins-Gerstl, N. Munro, M. Trucco, L. C. Brody and J. P. Landers, Genomics, 2000, 63, 25 CrossRef CAS.
- H. J. Tian, L. C. Brody and J. P. Landers, Genome Res., 2000, 10, 1403 CrossRef CAS.
- D. Schmalzing, A. Adourian, L. Koutny, L. Ziaugra, P. Matsudaira and D. Ehrlich, Anal. Chem., 1998, 70, 2303 CrossRef CAS.
- C. N. Hestekin, J. P. Jakupciak, T. N. Chiesl, C. W. Kan, C. D. O'Connell and A. E. Barron, Electrophoresis, 2006, 27, 3823 CrossRef CAS.
- G. Vahedi, C. Kaler and C. J. Backhouse, Electrophoresis, 2004, 25, 2346 CrossRef CAS.
- Y. Endo, L. Zhang, R. Katashima, M. Itakura, E. A. S. Doherty, A. E. Barron and Y. Baba, Electrophoresis, 2005, 26, 3380 CrossRef CAS.
- S. Stenirri, M. Cretich, I. Rech, A. Restelli, M. Ghioni, S. Cova, M. Ferrari, L. Cremonesi and M. Chiari, Electrophoresis, 2008, 29, 4972 CrossRef CAS.
- I. Rech, S. Cova, A. Restelli, M. Ghioni, M. Chiari and M. Cretich, Electrophoresis, 2006, 27, 3797 CrossRef CAS.
- H. Shadpour, H. Musyimi, J. Chen and S. A. Soper, J. Chromatogr., A, 2006, 1111, 238–251 CrossRef CAS.
- M. Zuborova, Z. Demianova, D. Kaniansky, M. Masar and B. Stanislawski, J. Chromatogr., A, 2003, 990, 179–188 CrossRef CAS.
- M. E. Cavalier, M. M. Davis and J. M. Croop, J. Pediatr. Hematol. Oncol., 2005, 27, 441 CrossRef.
| This journal is © The Royal Society of Chemistry 2012 |