 Open Access Article
Open Access ArticleSelf reporting RNA probes as an alternative to cleavable small molecule mass tags†
Jo-Anne
Riley
a,
Tom
Brown
a,
Nittaya
Gale
b,
Julie
Herniman
a and
G. John
Langley
*a
aChemistry, FNES, University of Southampton, Southampton, SO17 1BJ, UK. E-mail: gjl@soton.ac.uk; Tel: +44 (0)23 8059 2182
bATDBio LTD, University of Southampton, Southampton, SO17 1BJ, UK. E-mail: n.gale@soton.ac.uk; Fax: +44 (0)23 8059 2991; Tel: +44 (0)23 8059 6778
First published on 19th October 2012
Abstract
The large size of biological molecules such as proteins and oligonucleotides makes them inherently problematic to analyse and quantify directly by mass spectrometry. For these molecules, electrospray ionisation produces multiply charged species and associated alkali metal adducts which can reduce sensitivity and complicate quantification. Whereas time-of-flight mass analysers, often coupled to matrix-assisted laser desorption/ionisation, can have insufficient mass resolution to resolve these large molecules in the higher m/z range. This has led to the development of cleavable small molecule mass tag approaches for the indirect analysis of biomolecules such as proteins and oligonucleotides. Existing methodologies require the design and synthesis of a cleavable linker to join the biomolecule and the mass tag. Here, an alternative approach to small molecule mass tags is presented, which exploits the properties of the RNA molecule to afford self-reporting probes which can be easily synthesised using automated phosphoramidite chemistry. The sugar-phosphate backbone of RNA was used as a built-in enzyme cleavable linker and through the use of RNase digestion of bromine labelled oligonucleotides the observation of a range of small molecule mass tags by mass spectrometry is demonstrated. This study provides a proof-of-concept that RNase digestion can be used to produce labelled small molecule mass tags from oligonucleotide probes, thus eliminating the need for custom design and synthesis of a cleavable linker.
Introduction
Over the past four decades mass spectrometry has become an invaluable tool for the analysis of large biological molecules such as proteins and oligonucleotides. This is largely due to the advent of ionisation techniques capable of forming intact gas phase ions from biomolecules in solution; namely electrospray ionisation (ESI)1 and matrix-assisted laser desorption/ionisation (MALDI).2,3 However, these techniques have drawbacks when directly analysing large biomolecules, e.g. where electrospray ionisation produces a series of multiply charged ions as well as adducts with ubiquitous alkali metals such as sodium and potassium. These factors combine to dissipate the ion signal of one analyte across a number of m/z values, complicating the mass spectra, reducing sensitivity and hindering quantification. Although MALDI produces primarily singly charged ions, reproducibility can be poor due to formation of inhomogeneous matrix crystals with an uneven distribution of analyte across the sample spot. In addition, time-of-flight (TOF) mass analysers, commonly coupled to MALDI, can have insufficient mass resolution in the higher m/z range and so may be unable to distinguish between two ions of similar mass.To avoid these inherent problems of directly analysing large biomolecules, methods have been developed which use mass spectrometry for indirect analysis via measurement of small molecules. Peptide mass fingerprinting is a method of analysing proteins via mass spectral analysis of smaller characteristic peptides following an enzymatic digest.4–6 Observed peptide masses are searched against a list of theoretical digestion products of proteins on a database to identify the protein from which they originated. RNA mapping is an analogous technique for the analysis of RNA. Large RNA molecules, such as transfer RNAs or ribosomal RNAs, are digested with RNase enzymes. The masses of the digestion products are used to identify the original oligonucleotide and can locate and characterise post-translational modifications.7 Another method of indirect analysis of large biomolecules is via small molecule mass tags. The isotope-coded affinity tag (ICAT) method8 uses isotopically labelled mass tags covalently attached to proteins. Relative quantification of proteins from two different cell states can be achieved using a protein digest followed by mass spectral analysis of the mass tagged digest products. Tandem mass tags (TMTs)9 and isobaric tags for relative and absolute quantification (iTRAQ)10 use similar covalently attached isotopically labelled tags and digestion procedures. An MS/MS approach is then used to fragment the mass tagged peptides, simultaneously fragmenting the tags, for quantification, and the peptides, to produce sequence information.
Mass tags have also been used for the indirect analysis of oligonucleotides. Olejnik et al.11 used peptides as mass tags attached to oligonucleotide probes via a photocleavable linker to detect target DNA sequences immobilised on a solid support in a hybridisation assay. The laser pulse used in MALDI photocleaved the peptide mass tag from the probe, and this was followed by TOF MS analysis. Thompson et al.12 used a similar approach for the detection of target DNA sequences, however the mass tags were attached to the oligonucleotide probe via an ESI cleavable linker. Photocleavable mass tags have also been used for single nucleotide polymorphism (SNP) genotyping. The Masscode system13 uses small molecule mass tags attached via a photocleavable linker to an allele specific primer used in a polymerase chain reaction (PCR). The linker is cleaved by UV light and the tag then analysed by mass spectrometry. Hammond et al.14 also used photocleavable mass tags for SNP analysis. Primers were synthesised with a photocleavable, brominated, quaternised peptide mass tag. Following a single base extension reaction, the mass tag was cleaved by the MALDI laser pulse and analysed by TOF MS. A similar approach using brominated peptide mass tags attached via a photocleavable linker to a peptide nucleic acid detection probe for SNP genotyping by MALDI-TOF MS has also been reported.15
All of the indirect analysis approaches described above required the custom design and synthesis of a cleavable linker to join the large biomolecule to the small molecule mass tag, which often requires lengthy, non-automated solution phase synthesis. Ideally, the linker will give fast and highly efficient cleavage and be simple to synthesise. In addition, the chemistry required for conjugation to the biomolecule must be compatible with that of the biomolecule, the tag and the linker itself.
An alternative approach which uses mass spectrometry for indirect analysis via cleavable small molecule mass tags is presented here. This approach exploits the properties of the RNA molecule to provide the potential for a range of small molecule mass tags, whilst avoiding the need to design or synthesise a specific, new cleavable linker. RNase enzymes catalyse the hydrolysis of the phosphodiester backbone of RNA and can produce small molecules, such as mono, di- and trinucleotides from longer RNA probes. There are numerous types of RNase enzymes with different specificities, for example RNase A specifically cleaves at the 3′ side of cytidine and uridine in single stranded RNA,16 whereas RNase T1 specifically cleaves at the 3′ side of guanosine.17 Custom design of oligonucleotide sequences followed by digestion with a specific RNase enzyme affords products which can serve as cleavable small molecule mass tags for mass spectral analysis. Incorporation of atoms with distinctive isotope patterns, such as chlorine or bromine, used to label certain bases within an oligonucleotide probe leads to the production of labelled digestion products. This affords signals from the mass tags that are easily distinguishable from background signals, either by eye or by cluster pattern recognition software.
In this work, oligonucleotide sequences containing the modified base 5-bromouridine (UBr) were designed, synthesised by automated phosphoramidite chemistry, digested by RNase A and subsequently analysed by mass spectrometry. Initial experiments using RNA indicated that the specificity of RNase A was insufficient to guarantee cleavage only at the pyrimidine bases. Therefore, probes were redesigned as DNA–RNA chimeras with RNA bases only in positions where cleavage was desired. Observation of brominated mononucleotides, dinucleotides and trinucleotides provides a proof-of-concept. This will allow the future design of a range of probes which can be digested to produce a range of small molecule mass tags for the indirect analysis of large biomolecules.
Results
5-Bromouridine was used as the modified base in this study and therefore RNase A was chosen as the digestion enzyme. RNase A is an endonuclease which cleaves at the 3′ side of the pyrimidine bases, cytidine and uridine, and also therefore cleaves at the 3′ side of 5-bromouridine. The cleavage mechanism results in the formation of 3′ phosphate and 5′-OH oligonucleotides via a 2′, 3′ cyclic phosphate intermediate (Fig. 1).18 Six custom designed oligonucleotide (ONT) sequences used in this study along with their expected digestion products are shown in Table 1.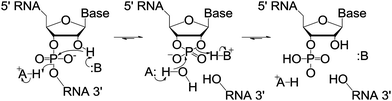 | ||
| Fig. 1 Cleavage mechanism of RNase A, where B is His12 and A is His119 (ref. 18). | ||
| ONT | Sequence | MW(mono) | Expected digestion products | MW(mono) |
|---|---|---|---|---|
| a UBr = 5-bromouridine, d = deoxynucleotide, p = 3′-phosphate. | ||||
| 1 | UCAAUBrUCAAUBrUCAAUBrUCAA | 6166.5 | Cp | 323.1 |
| Up | 324.0 | |||
| AA | 596.1 | |||
| AAUBrp | 1060.1 | |||
| 2 | UGACUBrUGACUBrUGACUBrUGAC | 6230.5 | Up | 324.0 |
| UBrp | 401.9 | |||
| GAC | 917.2 | |||
| GACp | 997.2 | |||
| 3 | dTdGCdAUBrdTdGCdAUBrdTdGCdAUBrdTdGCdA | 6094.6 | dTdGCp | 956.2 |
| dAUBrp | 715.0 | |||
| dA | 251.1 | |||
| 4 | dTCdAdAUBrdTCdAdAUBrdTCdAdAUBrdTCdAdA | 6030.7 | dTCp | 627.1 |
| dAdAUBrp | 1028.1 | |||
| dAdA | 564.2 | |||
| 5 | dTCdGdAUBrdTCdGdAUBrdTCdGdAUBrdTCdGdA | 6094.6 | dTCp | 627.1 |
| dGdAUBrp | 1044.1 | |||
| dGdA | 580.2 | |||
| 6 | dTCdGdGUBrdTCdGdGUBrdTCdGdGUBrdTCdGdG | 6158.6 | dTCp | 627.1 |
| dGdGUBrp | 1060.1 | |||
| dGdG | 596.1 | |||
Analysis of each ONT immediately prior to addition of RNase A confirmed that the oligonucleotides were intact at the beginning of the experiments (ESI, Fig. S1–S6†). Total ion current chromatograms (TICCs) and UV chromatograms of the digested assay samples showed no evidence of the intact oligonucleotides in the samples taken after 5 minutes and instead, the expected digestion products were observed. All expected species were observed by HPLC-ESI MS, except for 3′-OH mononucleotides from the 3′ terminus and by MALDI-TOF MS mononucleotides were generally not observed.
Negative ion ESI MS analysis of ONT 1 showed the expected brominated mass tag AAUBrp at m/z 1059.0, but also showed the production of the dinucleotide AUBrp at m/z 723.0. The peaks relating to this species in the UV chromatograms increased in area over time, as did the signal intensity of the ion in the mass spectra (ESI, Fig. S8†). This species results from overdigestion and non-specific cleavage after the adenosine base. Production of AUBrp from ONT 1 would become problematic if this sequence were to be used in a multiplexed system alongside a sequence which was designed to produce AUBrp as the mass tag because it could lead to a false positive result. Attempts to prevent overdigestion by using a ten-fold reduction of RNase A concentration were made, however whilst the extent of overdigestion was reduced, it was not eliminated (ESI, Fig. S9†). Therefore, to avoid cleavage at undesired sites, DNA–RNA chimeric oligonucleotide sequences were designed, where RNA pyrimidine bases were only located at the positions where RNase A cleavage was desired and all other bases were DNA.
HPLC-ESI MS analyses of ONTs 2, 3, 4, 5 and 6 digested with RNase A showed the expected brominated mass tags dUBrp, dAUBrp, dAdAUBrp, dGdAUBrp and dGdGUBrp, respectively, as singly (all mass tags) and doubly (trinucleotide mass tags) charged ions with the distinctive bromine isotope patterns (Fig. 2 and Table 2). The expected unlabelled products were also observed (Fig. 2(a) and S7†). Additionally, bromine labelled dinucleotides were observed upon digestion of ONTs 4, 5 and 6 designed to produce bromine labelled trinucleotides, (see Discussion below).
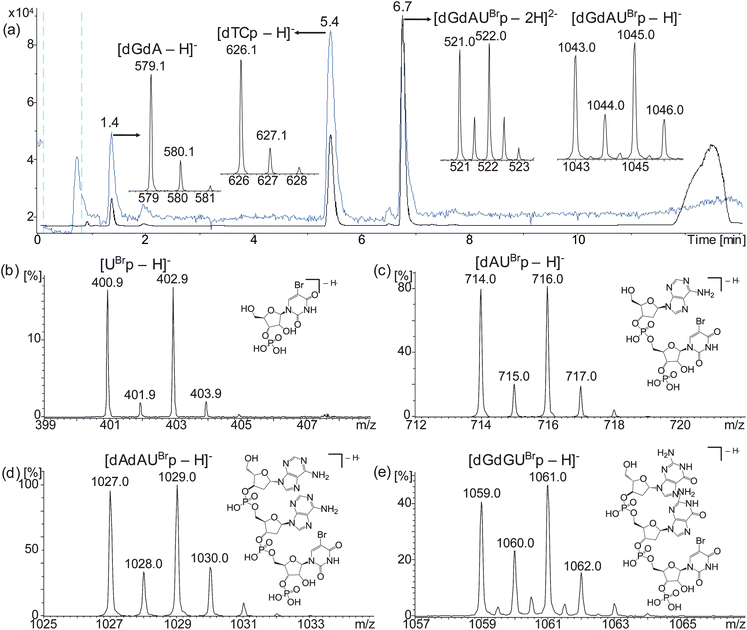 | ||
| Fig. 2 Representative HPLC-ESI MS analysis of ONT 5 sampled at 5 minutes showing TICC (blue), UV (black) and negative ion ESI mass spectra of observed digestion products (a) and negative ion ESI mass spectra of the brominated mass tags produced by digestion of ONTs 2 (b), 3 (c), 4 (d) and 6 (e). UBr = 5-bromouridine, d = deoxynucleotide, p = 3′-phosphate. | ||
| ONT | Expected mass tag | Calculated MW(mono) | ESI-MS observed m/z[M − H]− | MALDI-MS observed m/z[M + H]+ |
|---|---|---|---|---|
| a UBr = 5-bromouridine, d = deoxynucleotide, p = 3′-phosphate. | ||||
| 1 | AAUBrp | 1060.1 | 1059.0 | 1061.3 |
| 2 | UBrp | 401.6 | 400.9 | — |
| 3 | dAUBrp | 715.0 | 714.0 | 715.8 |
| 4 | dAdAUBrp | 1028.1 | 1027.0 | 1029.0 |
| 5 | dGdAUBrp | 1044.1 | 1043.0 | 1044.4 |
| 6 | dGdGUBrp | 1060.1 | 1059.0 | 1060.9 |
MALDI-TOF MS analyses of ONTs 3, 4, 5 and 6 digested with RNase A showed the expected brominated mass tags dAUBrp, dAdAUBrp, dGdAUBrp and dGdGUBrp, respectively, however the brominated mononucleotide UBrp produced by ONT 2 was not observed using this technique (Fig. 3 and Table 2). The expected unlabelled dinucleotides and trinucleotides were also observed by MALDI-TOF MS.
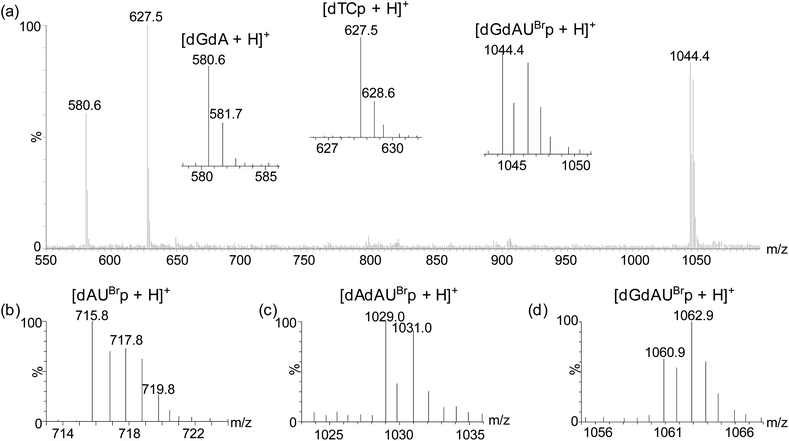 | ||
| Fig. 3 Representative MALDI-TOF MS analysis of ONT 5 sampled at 5 minutes showing positive ion mass spectra of observed digestion products (a) and positive ion MALDI-TOF mass spectra of the brominated mass tags produced by digestion of ONTs 3 (b), 4 (c) and 6 (d). | ||
Discussion
The use of MALDI-TOF MS provided a rapid method capable of simultaneously observing numerous digestion products. However, mononucleotides were not consistently observed under the conditions used and the bromine labelled mononucleotide was not observed using this technique. HPLC-ESI MS has the advantage of chromatographic separation, which acts to desalt the analytes and reduces the effect of ion suppression problems which may arise from the simultaneous analysis of ions when using MALDI-TOF MS.DNA bases lack the 2′ OH group required by the RNase mechanism to cleave the sugar-phosphate backbone. Therefore, the DNA–RNA chimeric sequences designed to produce bromine labelled trinucleotides should not be able to produce bromine labelled dinucleotides. Unfortunately, this was not the case and these species were still observed upon digestion of the chimeras, with their peak areas in the base peak chromatograms (BPCs) being typically less than 1% compared to those of the expected trinucleotides. However, the peak areas relating to the overdigestion products did not increase over time for the chimeric sequences as they did for the RNA sequences, as exemplified by the comparison of ONTs 1 and 4 (Fig. 4). It is therefore believed that the dinucleotides observed upon digestion of the chimeric sequences are a result of the normal RNase A digestion of the n-3 failure sequences which have the dAUBrp sequence at their 3′ terminus ( ESI, Fig. S10†).
 | ||
| Fig. 4 A graph showing the peak area of AUBrp and dAUBrp compared to AAUBrp and dAdAUBrp for ONTs 1 (RNA) and 4 (DNA–RNA chimera), respectively. Error bars show ±1 standard deviation from triplicate results. UBr = 5-bromouridine, d = deoxynucleotide, p = 3′-phosphate. | ||
Observation of the brominated digestion products UBrp, dAUBrp, dAdAUBrp, dGdAUBrp and dGdGUBrp by mass spectrometry provides a proof-of-concept that RNase digestion can be used to produce small molecule mass tags from DNA–RNA chimeric probes, eliminating the need for a synthetic cleavable linker. This indirect approach has the potential for the analysis of a variety of large biomolecules. For example, brominated nucleotide digestion products could form the signals of detection probes used in a hybridisation assay for the analysis of target oligonucleotide sequences (Fig. 5). Additionally, these chimeric probes could be conjugated to peptides or proteins for their indirect analysis.
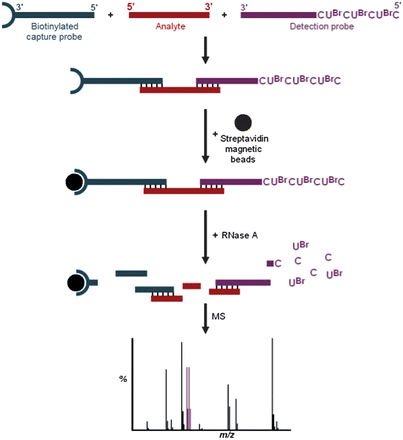 | ||
| Fig. 5 Example hybridization assay using mass spectral analysis of brominated RNase digestion products. | ||
This study has demonstrated the production and observation of five small molecule mass tags, however the approach has the potential for a greater degree of multiplexing via a number of methods. It would not be possible for more than one cytidine or uridine to be present in mass tags produced from RNase A digestion of RNA probes because cleavage would occur. The mass difference between cytidine and uridine of just 1 Da would also limit their concurrent use in a multiplexed system. However, through the use of DNA–RNA chimeras, inclusion of deoxycytidine (or deoxyuridine) and deoxythymidine would allow the range of mass tags to be extended to include dCUBrp, dTUBrp, dCdCUBrp, dTdTUBrp, dAdCUBrp, dAdTUBrp, dGdCUBrp, dGdTUBrp and dCdTUBrp as well as dGUBrp (or a series where dC is replaced by dU). An even wider range of mass tags could be produced by further modifying the bases to contain additional functional groups giving them additional mass. Furthermore, the use of additional isotope patterns, which could be achieved through the use of chlorine, multiple chlorine/bromine atoms and synthesis of bases containing custom isotope patterns, would further extend the multiplexing possibilities.
Experimental
Oligonucleotide synthesis and purification
ONTs synthesis was performed on an Applied Biosystems 394 automated DNA/RNA synthesiser. The phosphoramidite monomers for synthesis of the DNA–RNA chimeras were dmf-dG-CE, Bz-dA-CE, Bz-dC-CE, dT-CE, 5-Br-rU-CE and Ac-rC-CE and were purchased from Link Technologies (Lanarkshire, UK) and Applied Biosystems Ltd (Paisley, UK). All ONTs were synthesised on a 1.0 μmol phosphoramidite cycle with the following steps; acid catalysed detritylation, coupling, capping and iodine oxidation. Directly before use, β-cyanoethyl phosphoramidite monomers were dissolved in anhydrous acetonitrile to a 0.1 M concentration. Coupling time for the standard DNA monomers was 40 seconds and coupling time for the 5-Br-rU and rC was 600 seconds. Cleavage from the solid support in conjunction with exocyclic amino group deprotection was completed by exposing the solution to a mixture of aqueous ammonia–ethanol (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) for 2.5 hours at room temperature and then heated in sealed tube at 55 °C for 2 hours.
:1) for 2.5 hours at room temperature and then heated in sealed tube at 55 °C for 2 hours.
Purification of ONTs was achieved by reversed-phase HPLC using a Gilson system with a Phenomenex C8 column (10 μm, 10 mm × 250 mm). The following protocols were used: run time 20 min, flow rate 4 mL min−1, binary gradient: time in min. (% buffer B); 0 (0); 3 (0); 3.5 (5); 15 (45); 16 (100); 17 (100); 17.5 (0); 20 (0). Buffer A: 0.1 M triethylammonium acetate (TEAA) in water, pH 7.0, buffer B: 0.1 M TEAA in water–acetonitrile (1:1), pH 7.0. Elution of ONTs was monitored by UV absorption. ONTs were then desalted using NAP-25 and NAP-10 Sephadex columns (G-25, GE Healthcare), aliquoted into Eppendorf tubes and stored at −20 °C.
Digestion assay
For digestion, an ONT was dissolved in 220 mM ammonium acetate buffer prepared with sterile water and heated to 37 °C.19 RNase A, dissolved in the ammonium acetate buffer, was added and the assay kept at 37 °C. A total volume of approximately 45 μL was used with an RNA concentration of 2 μg μL−1 and 0.1 Kunitz units20 of RNase A per μg RNA. A control sample of RNA in ammonium acetate buffer was also kept at 37 °C for the duration of the experiment. Samples were taken from the assay and the control sample at intervals of 5 minutes, 2, 4 and 24 hours and were analysed by MALDI-TOF MS and HPLC-ESI MS.HPLC-ESI MS
Samples were prepared for HPLC-ESI MS analysis by adding 1 μL of assay solution to 19 μL of sterile water in a plastic vial. HPLC separation was performed using a Dionex UltiMate 3000 liquid chromatography system with a quaternary solvent delivery system, UV/visible detector, heated column compartment and chilled autosampler (Dionex, Hemel Hempstead, UK). An Acquity UPLC BEH C18 column (1.7 μm, 1 mm × 100 mm, Waters, Milford, MA, USA) was used for separation. The column temperature was set to 40 °C and UV absorbance was measured at 290 nm. A binary gradient solvent system was used with mobile phase A consisting of 10 mM TEAA and 100 mM hexafluoroisopropanol (HFIP) in water and mobile phase B consisting of 20 mM TEAA in acetonitrile. For analysis of intact oligonucleotides, mobile phase B was at 5% for 1 minute, then increased to 40% over 14 minutes and then returned to 5% until 18 minutes. For analysis of digested samples, mobile phase B was at 1% for 1 minute, then increased to 20% over 7 minutes, then increased to 40% over 2 minutes and then returned to 1% until 13 minutes. A flow rate of 100 μL min−1 and a 2 μL injection volume were used. Negative ion ESI data were acquired using a MicrOTOF (Bruker, Bremen, Germany) mass spectrometer over the m/z range 250–3500. Data were analysed using Data Analysis™ software v4.0.MALDI-TOF MS
Samples were prepared for MALDI-TOF MS analysis by spotting 1 μL Dowex H+ ion exchange beads onto a MALDI plate for desalting,21 followed by 1 μL of assay solution and 1 μL of matrix and mixing within the pipette tip. A 10:1 mixture of 3-hydroxypicolinic acid (3-HPA)–picolinic acid (PA) in 1:1 acetonitrile–water was used as the matrix. The spot was left to dry under a stream of warm air. Positive ion data were acquired using a Micromass TofSpec 2E MALDI-TOF mass spectrometer (Manchester, UK) in reflectron mode with an acceleration voltage of 20 kV and a pulse voltage of 3 kV. A nitrogen laser was used operating at a wavelength of 337 nm with a pulse duration of 5 ns. Each spectrum acquired was a summation of 5 laser shots and a total of 33 spectra were acquired by sampling in a spiral pattern around the sample spot. Data were analysed using MassLynx™ software v4.1.
Conclusions
An alternative approach to the indirect analysis of large biological molecules via small molecule mass tags using mass spectrometry has been demonstrated. The sugar-phosphate backbone of the RNA molecule was successfully used as a built-in enzyme cleavable linker and RNase A digestion of custom designed oligonucleotides containing 5-bromouridine was found to produce bromine labelled mono-, di- and trinucleotide small molecule mass tags. Although non-specific cleavage after purine bases was observed upon RNase A digestion of RNA, redesign of the probes as DNA–RNA chimeras circumvented this problem. Observation of the brominated digestion products UBrp, dAUBrp, dAdAUBrp, dGdAUBrp and dGdGUBrp by HPLC-ESI MS and MALDI-TOF MS provides a proof-of-concept that RNase digestion can be used to produce a range of small molecule mass tags with a high degree of multiplexing potential. This approach will allow oligonucleotides to be used as the detection probe in a hybridisation assay, the enzyme cleavable linker and the small molecule mass tag, i.e. they are multi-functional self-reporting probes. They can be easily synthesised by well established, automated phosphoramidite chemistry which eliminates the need for custom design of a synthetic cleavable linker.Acknowledgements
The authors thank BBSRC/Pfizer for the PhD CASE Award and ATDBio for oligonucleotide synthesis.Notes and references
- J. B. Fenn, M. Mann, C. K. Meng, S. F. Wong and C. M. Whitehouse, Science, 1989, 246, 64–71 CAS.
- M. Karas, D. Bachmann, U. Bahr and F. Hillenkamp, Int. J. Mass Spectrom. Ion Processes, 1987, 78, 53–68 CrossRef CAS.
- K. Tanaka, Y. Ido, S. Akita, Y. Yoshida and T. Yoshida, Proceedings of the Second Japan-China Joint Symposium on Mass Spectrometry, 1987 Search PubMed.
- J. R. Yates, S. Speicher, P. R. Griffin and T. Hunkapiller, Anal. Biochem., 1993, 214, 397–408 CrossRef CAS.
- W. J. Henzel, T. M. Billeci, J. T. Stults, S. C. Wong, C. Grimley and C. Watanabe, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 5011–5015 CrossRef CAS.
- P. James, M. Quadroni, E. Carafoli and G. Gonnet, Biochem. Biophys. Res. Commun., 1993, 195, 58–64 CrossRef CAS.
- J. A. Kowalak, S. C. Pomerantz, P. F. Crain and J. A. McCloskey, Nucleic Acids Res., 1993, 21, 4577–4585 CrossRef CAS.
- S. P. Gygi, B. Rist, S. A. Gerber, F. Turecek, M. H. Gelb and R. Aebersold, Nat. Biotechnol., 1999, 17, 994–999 CrossRef CAS.
- A. Thompson, J. Schafer, K. Kuhn, S. Kienle, J. Schwarz, G. Schmidt, T. Neumann and C. Hamon, Anal. Chem., 2003, 75, 1895–1904 CrossRef CAS.
- P. L. Ross, Y. L. N. Huang, J. N. Marchese, B. Williamson, K. Parker, S. Hattan, N. Khainovski, S. Pillai, S. Dey, S. Daniels, S. Purkayastha, P. Juhasz, S. Martin, M. Bartlet-Jones, F. He, A. Jacobson and D. J. Pappin, Mol. Cell. Proteomics, 2004, 3, 1154–1169 CAS.
- J. Olejnik, H. C. Ludemann, E. Krzymanska-Olejnik, S. Berkenkamp, F. Hillenkamp and K. J. Rothschild, Nucleic Acids Res., 1999, 27, 4626–4631 CrossRef CAS.
- A. Thompson, M. Prescott, N. Chelebi, J. Smith, T. Brown and G. Schmidt, Nucleic Acids Res., 2007, 35, e28 CrossRef.
- M. Kokoris, K. Dix, K. Moynihan, J. Mathis, B. Erwin, P. Grass, B. Hines and A. Duesterhoeft, Mol. Diagn., 2000, 5, 329–340 CAS.
- N. Hammond, P. Koumi, G. J. Langley, A. Lowe and T. Brown, Org. Biomol. Chem., 2007, 5, 1878–1885 CAS.
- R. Ball, Ph.D. thesis, University of Southampton, 2005.
- E. Volkin and W. E. Cohn, J. Biol. Chem., 1953, 205, 767–782 CAS.
- K. Sato and F. Egami, J. Biochem., 1957, 44, 753–767 CAS.
- J. E. Thompson, F. D. Venegas and R. T. Raines, Biochemistry, 1994, 33, 7408–7414 CrossRef CAS.
- M. Hossain and P. A. Limbach, Anal. Bioanal. Chem., 2009, 394, 1125–1135 CrossRef CAS.
- M. Kunitz, J. Biol. Chem., 1946, 164, 563–568 CAS.
- G. J. Langley, J. M. Herniman, N. L. Davies and T. Brown, Rapid Commun. Mass Spectrom., 1999, 13, 1717–1723 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: HPLC-ESI MS analyses of intact oligonucleotides and additional overdigestion data. See DOI: 10.1039/c2an36086a |
| This journal is © The Royal Society of Chemistry 2012 |