Label-free electrical detection of pyrophosphate generated from DNA polymerase reactions on field-effect devices†
Grace M.
Credo
*a,
Xing
Su
a,
Kai
Wu
a,
Oguz H.
Elibol
ab,
David J.
Liu
a,
Bobby
Reddy
Jr.
c,
Ta-Wei
Tsai
a,
Brian R.
Dorvel
c,
Jonathan S.
Daniels
a,
Rashid
Bashir
*c and
Madoo
Varma
*a
aIntegrated Biosystems Lab, Intel Labs, Intel Corporation, 2200 Mission College Blvd., Santa Clara, CA 95054, USA. E-mail: grace.m.credo@intel.com; madoo.varma@intel.com
bBirck Nanotechnology Center and School of Electrical and Computer Engineering, Purdue University, West Lafayette, IN 47907, USA
cMicro and Nanotechnology Laboratory and Departments of Electrical and Computer Engineering and Bioengineering, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA. E-mail: rbashir@uiuc.edu
First published on 19th January 2012
Abstract
We introduce a label-free approach for sensing polymerase reactions on deoxyribonucleic acid (DNA) using a chelator-modified silicon-on-insulator field-effect transistor (SOI-FET) that exhibits selective and reversible electrical response to pyrophosphate anions. The chemical modification of the sensor surface was designed to include rolling-circle amplification (RCA) DNA colonies for locally enhanced pyrophosphate (PPi) signal generation and sensors with immobilized chelators for capture and surface-sensitive detection of diffusible reaction by-products. While detecting arrays of enzymatic base incorporation reactions is typically accomplished using optical fluorescence or chemiluminescence techniques, our results suggest that it is possible to develop scalable and portable PPi-specific sensors and platforms for broad biomedical applications such as DNA sequencing and microbe detection using surface-sensitive electrical readout techniques.
Introduction
Inorganic pyrophosphate (PPi) is a reaction byproduct of nucleotide base incorporation reactions catalyzed by DNA or RNA polymerases. These reactions are critically important in the biological processes of living systems.1PPi or diphosphate (P2O74−) is a small, negatively charged molecule which is highly mobile. With appropriate enzymes and sufficient nucleotides in solution, PPi and protons can be generated repeatedly for a single reversible nucleotide base addition at the DNA polymerase binding site, generating multiple reaction product molecules, and greatly amplifying the signal to noise ratio of detection techniques targeting PPi.2,3Detection of DNA polymerase activity and subsequent PPi release is typically detected using optical techniques, such as chemiluminescence.4 For example, luciferase-based PPi detection has been used for bacterial detection and DNA sequencing-by-synthesis applications. Non-enzymatic PPi detection based on fluorescence- and absorption-based techniques using PPi chelators has been reported as well.2 Extending chelation-based sensing to surface-sensitive electrical detection requires a chelator compatible with surface immobilization and selective to the target analyte. We have previously reported the synthesis of a surface-immobilizable chelator based on di-(2-picolyl) amine (DPA) which has demonstrated strong binding affinity to PPi and demonstrated reversible SOI-FET sensing of PPi standard solutions.5Key advantages of electronic biosensing compared to optically-based biological sensing include highly scalable semiconductor manufacturing technology, simplified instrumentation due to the elimination of bulky optical components such as microscopes and lasers, and the potential for seamless sample-to-answer integration with functions such as data acquisition and database mining co-located on the same chip.6,7 To this end, scalable semiconductor manufacturing techniques have been used to produce sensor arrays for the electronic measurement of biochemical reactions in a highly parallel fashion.8–11
For surface-sensitive techniques, such as electronic signal transduction, the ability to detect the target event on the sensor is a function of complex relationships among the chemical properties of the sensor surface, the material properties of the constructed device, control of chemical interactions at the sensor surface, and transport effects in the surrounding solution. Chemical modifications of sensor surfaces can introduce custom-designed affinity binding sites which can either tune device response to specific biochemical interactions, introducing characteristic binding kinetics to analyte interactions with the sensor surface or can minimize the probability of non-specific binding.12–15 Magnetic beads have been used to separate DNA immobilization and amplification from active optical or electrical sensor devices, avoiding sensor exposure to lengthy DNA preparation procedures as well as increasing local analyte concentrations in order facilitate signal detection.11Buffer conditions such as pH, ionic strength, and surfactants offer an additional measure of control, either facilitating or hindering the target chemical interaction.16
A number of reports describe the charge-based response of field effect sensors for label-free, electronic detection of deoxyribonucleic acid (DNA) where probe oligonucleotides in solution bind to target strands in a sensor array, increasing negative charge at the sensor surface.17–19 However, the hybridization of partially mismatched target DNA to DNA oligomer probes on the surface is difficult to eliminate, requiring control of salt concentrations, temperatures, and pH near the sensor surface.20 As sensor areas are scaled to increase the density of sensor arrays for massively parallel measurements, decreasing signal intensity approaching noise levels and nonspecific chemical interactions causing misleading false positives can be exacerbated.21,22 In addition, hybridization-based methods require prior knowledge of the target DNA sequences so that the corresponding probes are immobilized on the sensor surface before the surface assay. The use of field effect sensors for label-free detection of DNA using base extension chemistry, which is a more specific chemical interaction than hybridization,23,24 has been less common but recent developments have shown the promise of this approach. For example, the detection of transient signal caused by the differential diffusion of highly mobile protons generated by the DNA base incorporation reaction is the basis of semiconductor-based electronic sensing of DNA sequencing-by-synthesis reactions from functionalized beads placed near the transducer surface.11
In this work, we describe the chemical modification of SOI-FET devices with both chelator and DNA colonies and demonstrate specific and static (non-transient) electrical response to DNA polymerase reactions in bulk solution and on the device surface using pyrophosphate as the target signaling molecule. A chip containing multiple devices was functionalized with both DNA on the surface and chelator molecules to capture pyrophosphates produced from the enzymatic incorporation reaction of a matching DNA base, as depicted in Fig. 1a. The chelator molecules were designed for the specific binding of pyrophosphate released from DNA polymerase-catalyzed base incorporation reactions, as depicted in Fig. 1b and 1c. As shown in Fig. 1d, the bidentate chelator complexes with zinc ions in solution and binds to the diphosphate target molecule. We used rolling circle amplification (RCA) of immobilized DNA in order to increase the number of parallel reactions occurring at each DNA binding site.25,26 The characteristic I–V transfer curve for the sensor was expected to remain unchanged unless PPi attached to the chelator, changing the source–drain current of the FET at a given back-gate potential. Based on charge effects alone, the interaction of the negatively charged PPi with the unmodified surface of a p-type accumulation mode SiO2/Si nanoplate SOI-FET device was expected to increase the number of carriers in the channel, increasing the drain current at a fixed potential and shifting the threshold voltage of the device more positive.27 However, the interaction of the pyrophosphate with the field-effect sensor occurs at the terminal end of a multi-component chemical surface modification with the device response to PPi binding a cumulative result of complex interactions and competing effects.
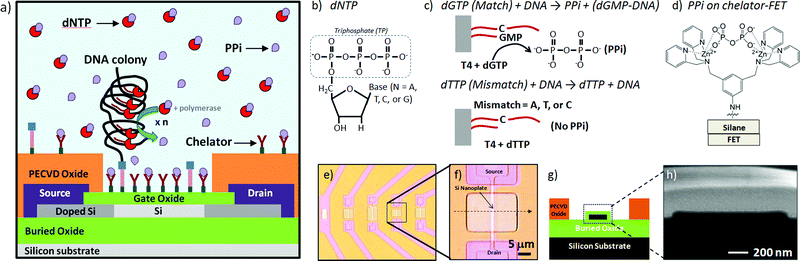 | ||
| Fig. 1 Pyrophosphate-generating DNA synthesis reactions and capture of pyrophosphate by chelators on an FET surface: a) the device is functionalized with both amplified DNA on the surface in the form of DNA colonies and chelator groups to capture pyrophosphates produced from the enzymatic incorporation reaction of a matching DNA base, b) generic DNA nucleotide triphosphate (dNTP) base structure, c) diphosphate (PPi) generated from a T4 polymerase-catalyzed dG monophosphate incorporation compared to control reaction, generating no PPi, d) chemical structure of PPi attached to chelator on modified FET surface, e) optical microscopy images of low density SOI nanoplate FET array, f) image of one of the devices used in these experiments, g) cross-section schematic of Si nanoplate FET device, and h) high resolution SEM image of ∼25 nm Si nanoplate cross-section of same device shown in (f). | ||
Materials and methods
Reagents
3-aminopropyltriethoxysilane (APTES) was obtained from Gelest. Glutaraldehyde (70%), water soluble 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide (EDC), and 200 proof anhydrous ethanol were obtained from Sigma. Reagents were diluted to the indicated concentrations in the following procedures, but otherwise were used as received. High resistivity water (18.2 MΩ) used for rinsing or diliutions was obtained from a Barnstead Nanopure purification system. Reducing agent sodium triacetoxyborohydride (STAB) was obtained from BASF and diluted in water as indicated. Polyethylene glycol (PEG, MW = 44) linker and blocker molecules amino-dPEG™8-acid (H2N-PEG8-COOH), amino-dPEG™12-acid (H2N-PEG12-COOH), and amino-dPEG™4-alcohol (H2N-PEG4-OH) were obtained from Quanta Biodesign Ltd. and diluted in water or buffer, as indicated. The synthesis and storage of the amine-functionalized chelator designed for PPi capture was described in a separate publication from our group.5Buffers used in these procedures included 1x phosphate buffered saline (PBS) at pH 8, 0.1 M 2-(N-morpholino)ethanesulfonic acid (MES) buffer at pH 5, and 0.2 M sodium borate buffer at pH 8. For rinsing non-specifically bound components such as avidin and storage of DNA colonies attached to device surfaces, we typically used a custom 1x ‘TEST’ (Tris, EDTA, Salt, Triton) buffer with composition 10 mM TrisHCl at pH 8, 1 mM EDTA, 50 mM NaCl and 0.01% Triton X-100. In addition we typically used a Tris-based buffer with composition 6.25 mM NaCl, 1.25 mM MgCl2,10 mM Tris pH 8 and 1 mM DTT as a blank measurement control before and after polymerase reactions.Immunopure avidin was obtained from Pierce and diluted in 1x PBS buffer. Unless otherwise specified in the RCA DNA preparation section, all DNA oligonucleotides were synthesized by IDT (Integrated DNA Technologies). Taq DNA polymerase was obtained from Applied Biosystems. The kits used for DNA purification were from Qiagen. Phi29 and buffers used with Phi29 were obtained from New England Biolabs (NEB).
Devices and measurement apparatus
The nanoplate SiO2/Si field effect devices were fabricated using standard lithographic and microfabrication techniques as previously described.28Optical microscopy images (Fig. 1e and 1f), a nanoplate cross-section diagram (Fig. 1g) and a high resolution scanning electron microscopy (SEM, Fig. 1h) cross-section image of devices used in this study are presented in Fig. 1. Briefly, the silicon field effect devices used to electrically detect PPi were fabricated using standard microfabrication techniques from a silicon-on-insulator (SOI) wafer (SOITEC). The FET devices had active areas with 1–2 μm widths and variable lengths: 10, 15 or 20 μm. The majority of the results reported here were obtained on devices with 1 (or 2) × 20 μm active areas. Full details of the device response to pH and other surface modifications are described in previous publications from the Bashir group.29Devices were mounted on a probe station (Signatone) and characterized using a Keithley Semiconductor Characterization System (SCS) Model 4200. Devices were connected to source, drain, backgate and top-gate (fluid-gate) voltage sources. Field effect devices used for the detection were operated in accumulation mode and the source–drain current was monitored as a function of the backgate bias (Vbg = −10 V to 2 V) at a fixed source–drain potential (Vsd = 100 and 30 mV). The I–V transfer curves reported in this work were collected with Vsd = 30 mV and Vsd = 100 mV, but all data presented in this work was collected with Vsd = 100 mV. Each device used in pyrosphosphate detection measurements was characterized in a variety of control conditions, including dry, in buffer with chelator-modified surface, and in buffer with unmodified surface. Some examples are presented in Fig. 3I–V transfer curve graph. A probe station enclosure protected the devices from light, sound and physical vibration during measurements.
For device measurements in solution, polydimethylsiloxane (PDMS, Dow Sylgard) was used to create a small well around many devices. These devices were then probed serially on a Signatone probe station. To this was added 2 to 3 μL of the indicated solutions, such as control buffer (1x PBS), control buffer with zinc (1x PBS with Zn(NO3)2, PPi in buffer with zinc, and control buffer with Zn2+ after an incubation and rinse in 0.1M acetic acid to remove Zn2+ complexed to the chelator and any PPi bound to the chelator coordination sites.
Silane modification
An overview of the initial surface modification is illustrated in Scheme 1, Steps 1 and 2. Substrates were cleaned in fresh, hot piranha (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 hydrogen peroxide:sulfuric acid) for at least 30 min. Caution: Piranha solution is highly corrosive and should be used with extreme caution. Following piranha cleaning, samples were rinsed thoroughly in water and dried in flowing nitrogen gas. Substrates were added to anhydrous ethanol with 1% 3-aminopropyltriethoxysilane (APTES). Then water was added to this solution at a final concentration of 2% by volume and substrates were incubated at room temperature for less than 10 min for monolayer silane deposition. Incubation times much longer than this often resulted in multilayer rather than monolayer formation, as measured by ellipsometry and AFM. After silane deposition, the chips were rinsed with ethanol and water, dried under nitrogen, and annealed for 10 min at 110 °C in order to promote cross-linking of the silane monolayer. For further functionalization, the amine-functionalized chips were immersed in a well-mixed solution of 2% glutaraldehyde in 1:1 ethanol:water. After 2 h, the chips were rinsed with copious amounts of water to remove non-specifically bound material on the surface and dried under nitrogen.
:3 hydrogen peroxide:sulfuric acid) for at least 30 min. Caution: Piranha solution is highly corrosive and should be used with extreme caution. Following piranha cleaning, samples were rinsed thoroughly in water and dried in flowing nitrogen gas. Substrates were added to anhydrous ethanol with 1% 3-aminopropyltriethoxysilane (APTES). Then water was added to this solution at a final concentration of 2% by volume and substrates were incubated at room temperature for less than 10 min for monolayer silane deposition. Incubation times much longer than this often resulted in multilayer rather than monolayer formation, as measured by ellipsometry and AFM. After silane deposition, the chips were rinsed with ethanol and water, dried under nitrogen, and annealed for 10 min at 110 °C in order to promote cross-linking of the silane monolayer. For further functionalization, the amine-functionalized chips were immersed in a well-mixed solution of 2% glutaraldehyde in 1:1 ethanol:water. After 2 h, the chips were rinsed with copious amounts of water to remove non-specifically bound material on the surface and dried under nitrogen.
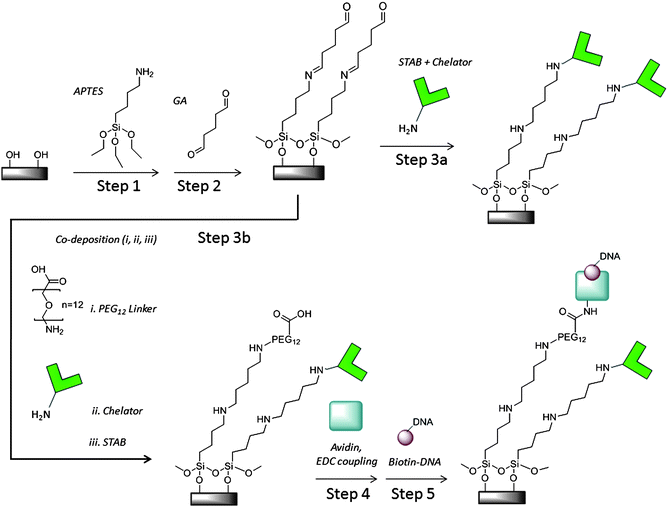 | ||
| Scheme 1 Pyrophosphate chelator and DNA colony silicon oxide surface modification | ||
Chelator modification
The PPi specificity and solution binding characteristics of the recently developed chelator have been previously reported.5 The chelator only modification step is depicted in Scheme 1, Step 3a. To attach amine-functionalized chelator molecules covalently on surfaces previously modified with glutaraldehyde, the surface was incubated in a solution of 1 mM chelator solution in 100 mM sodium borate buffer (pH 8), forming a Schiff base. After an hour, an equal volume of ∼0.2 mM sodium triacetoxyborohydride (STAB) solution in water was added to the chelator solution on chip and incubated overnight for reductive amination. Then the device surface was washed with 10 mM sodium borate buffer (pH 8) and with Tris control buffer (6.25 mM NaCl, 1.25 mM MgCl2,10 mM Tris pH 8 and 1 mM DTT) and stored in control buffer prior to experiments.The co-deposition of chelator with PEG linkers that serve as avidin attachment sites is depicted in Scheme 1, Step 3b. The attachment chemistry is similar to that for chelator only. Typically, the chelator and PEG linker, H2N-PEG12-COOH (Quanta Biodesign Ltd.), solutions were mixed in a 1:5 ratio, approximately 200 uM to 1 mM in 100 mM borate buffer and deposited on silicon pieces or devices. After an hour, ∼0.2 mM STAB solution was added to the chip surfaces and incubated overnight. Then the device surface was washed as described in the previous section and stored in the control buffer before measurements. For surface characterization with ellipsometry, AFM or TOF-SIMS, devices and silicon pieces were dried under nitrogen gas and stored in vacuum packed containers until analysis. Devices used for electrical measurements were modified in parallel.
Dipole moment calculations
Molecular dipole moments were calculated using the GAMESS computational chemistry package in Chem3D Pro using the RHF/3-21G level of theory and MM2 energy minimization and these results are presented in the ESI section.†Avidin modification
Prior to DNA exposure, avidin was attached to carboxylic acid terminated PEGn=8 or 12 linkers on the surface as shown in Scheme 1, Step 4. The carboxylic acid terminated linkers were activated by adding 0.1 mM EDC in MES pH 5 to the substrate surface. After a 30 min incubation, an equal volume of 50 μg mL−1 avidin in 1x PBS was added to the surface followed by incubation for 2 h at room temperature. Free avidin was removed by washing the wells 5 times with 1x TEST buffer (10 mM TrisHCl, pH 8, 1 mM EDTA, 50 mM NaCl and 0.01% Triton X-100). Similar procedures have been reported to control avidin orientation on surfaces and bind one biotinylated molecule per avidin binding site.30DNA preparation and surface attachment
The general scheme for DNA attachment on FET surfaces is depicted in Scheme 1, Step 5 and in Fig. 2a. Circular DNA was attached to the surface and one strand of the circular DNA was amplified in order to produce multiple copies of the same sequence on individual binding sites using a technique known as rolling circle amplification (RCA).25 Each DNA colony formed in this manner was a concatemer of a single stranded DNA (ssDNA) formed by amplifying one strand of a circular, doubled stranded DNA (dsDNA). For the surface-immobilized RCA-DNA used in this work, we used PCR to amplify a 260 bp fragment of the pUC19 plasmid using an upstream primer of 5′- pCTGCAATGATACCGCGAGACCCA-3′ and a downstream primer of 5′-pCCTTGATCGTTGGGAACCGGAG-3′. To ensure efficient ligation, purified PCR DNA was treated with Taq DNA polymerase (0.1 U/μl) in the presence of 500 μM dATP for 30 min. This treatment resulted in a single base A protruding from the 3′ end of each DNA strand (A-tail). The treated DNA was purified and quantified using UV absorption (Nanodrop) before ligation. Although the results described in this work were from PCR amplified DNA, we have also demonstrated RCA DNA colony formation on silicon oxide test surfaces and devices from total genomic DNA in parallel work.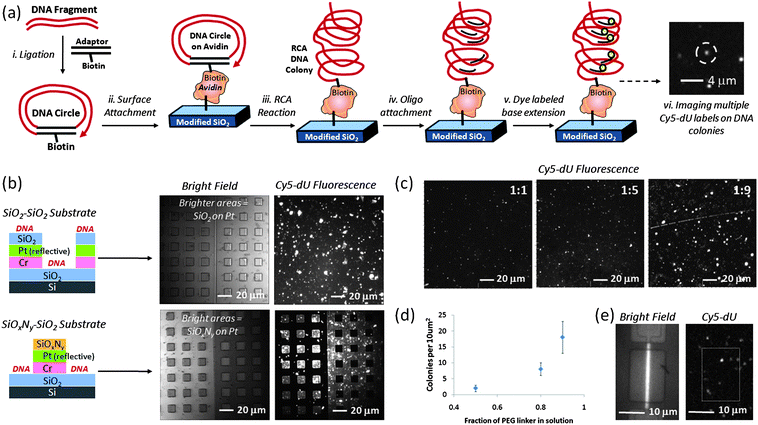 | ||
| Fig. 2
DNA colony formation process and characterization on surface: a) schematic of DNA sample preparation, colony formation and Cy5 dye labeling, b) relatively high density DNA colony modification of selectively patterned test substrates using APTES + GA + linker-only + avidin + biotinylated DNA attachment chemistry on silicon oxide, comparing coverage of fluorescently labeled DNA colonies on silicon oxide surfaces with coverage on the relatively dark silicon oxynitride surfaces, as labeled, c) adjusting DNA colony coverage on flat silicon oxide test substrates by changing the ratio of chelator to PEG-based DNA linker in reaction solution (1:1, 1:5, or 1:9), d) number of DNA colonies for every10 μm2silicon oxide substrate surface as function of PEG linker fraction, e) white light image (left) of representative silicon nanoplate SOI-FET device and corresponding fluorescence image of Cy5-labeled DNA colonies (right) using 1:5 chelator to PEG-linker deposition conditions. | ||
In principle, any DNA fragment with phosphate groups at the 5′ ends and hydroxyl group at the 3′ ends can be circularized by the following ligation procedure. A multi-purpose adaptor was assembled from a set of five DNA oligonucleotides present at equimolar ratio (1:1:1:1:1): RC1 (5′-pAGCTCGGCGGCCGCTTAAGT-3′), RC2Tb (5′-biotin-spacer-CTCCTATCACTTAAGCGGCCGCCGAGCTT-3′), RC3T (5′-pACGTCCGTACGTTCGGAACCT-3′), RC4 (5′-pGGTTCCGAACGTACGGACGTCCAGCTGAG-3′, 3′ locked nucleotide, resistant to 3′ → 5′ exonuclease), and RC5 (5′-pGATAGGAGATCTCAGCTGG-3′). The oligonucletide sequence information is also tabulated in the ESI section.† The oligo mixture was stored at −20 °C before use. The adaptor was designed so that after ligation each 5′ end was phosphorylated and the 3′ end had a single base T (T-tail). In the middle of the adaptor, there was a gap and the 5′ terminus of the gap was tagged with biotin and the 3′ terminus in the gap was rendered resistant to exonuclease digestion by use of a locked nucleotide at the −1 position. To form a circular DNA structure presented in Scheme 2, the A-tailed target DNA and the T-tailed adaptor DNA were mixed at equimolar ratio (1:1), typically 0.1–0.5 μM in a 50 μl ligation reaction. After ligation with T4 DNA ligase (0.2 U/uL) at 16 °C overnight, T4 DNA polymerase (10 U) was added to the sample directly to digest non-circular DNA molecules at 37 °C for 30 min. Purified DNA was quantified based on UV absorption.
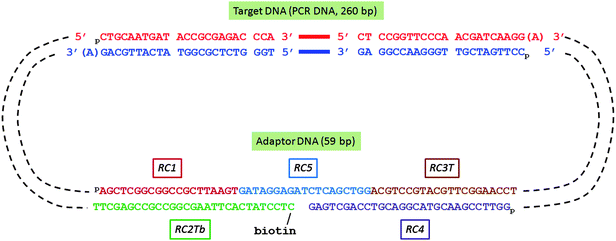 | ||
| Scheme 2 DNA oligonucleotides assembled in biotinylated DNA circle for Rolling Circle Amplification (RCA) | ||
Circularized DNA (biotin-functionalized) was diluted in 1x TEST buffer to a concentration of ∼107 copies/μL. In order to confine the solution to the FET device chip, a 2 mm × 1 mm custom PDMS well was trimmed to fit inside the pad area of the 3 mm × 5 mm chips. The diluted biotinylated DNA samples were incubated with the chelator with avidin on linker surface for at least 2 h, as depicted in Scheme 1, Step 5. Free DNA molecules were then removed by washing the wells 5 times with 1x TEST buffer.
Before rolling circle amplification (RCA) amplification, the wells were conditioned with 1x Phi29 buffer (NEB). For RCA amplification, about 10 μL of the reaction solution (1x Phi29 buffer, 200 μM dNTPs, and 0.5 U/μl Phi29 DNA polymerase) was added to each surface followed by incubation at 30 °C for at least 4 h or overnight to increase the number of DNA fragment copies per colony. Surface reactions were stopped by removing the reaction solution and washing the wells several times with 1x TEST buffer.
Surface analysis tools
The thickness of deposited films was measured using a Variable Angle Spectroscopic Ellipsometer (M2000FI VASE, J.A. Woollam, Lincoln, NE) scanning 685 wavelengths between 240 nm and 1100 nm at 65°, 70° and 75°.31 A Cauchy layer was used to model the organic monolayer on a surface. When possible, measurements were made on the same thermally oxidized silicon substrates before and after surface modification.The morphology of the sample surfaces was observed by AFM, as in Fig. 4. AFM was performed using a Dimension V Atomic Force Microscope (Veeco, Santa Barbara, CA). Scan rate was set to 1 Hz and the scan area was set between 1 μm2 and 100 μm2. TESPA silicon tips (Veeco) with 20–80 N m−1 stiffness and response frequency of ∼250–300 kHz were used. The instrument was operated in tapping mode. Images were flattened to adjust the plane and to remove scan lines. The height scale was adjusted to 15 nm. Feedback controls such as integral gain, proportional gain and amplitude setpoint were modulated in real time as the image was being generated in order to optimize image quality. Integral and proportional gains were typically set between 0.09 and 1.4.
Fluorescence images (Fig. 2 and 4) of Cy5-dUTP labeled RCA DNA colonies on substrates were obtained on a custom Nikon Eclipse ME600 microscope using a 10x or 50x Nikon objective with an EXFO X-Cite mercury halide lamp, a Cy-5 compatible Omega Optical filter cube, and an Andor iXon+ electron-multiplying (EM) CCD camera. Custom Andor software was used to process and adjust images. Acquisition times were typically 1s.
DNA polymerase reactions
The general scheme for PPi detection from DNA polymerase reactions conducted off-chip (ex situ) in a reaction tube and DNA polymerase reactions performed on surface-immobilized DNA colonies on FET surfaces (in situ) is depicted in Fig. 5a.For ex situ T4 polymerase reactions, the control buffer (T4 buffer) consisted of 10mM Tris pH 8 with 6.25 mM NaCl, 1.25 mM MgCl2, and 1mM DTT. The negative control solution consisted of the T4 buffer solution with 0.2 mM dTTP (mismatch base), 0.1 Unit T4 polymerase, 0.05 μM ‘DNA2’ hairpin DNA (5′-/5amMC6/GTC GCG CAA AAA TAC CTA GTC G + AC GTG GTC CTT/iBiodT/TT GG + A CCA CGT CGA CT + A G-3′). The positive control solution consisted of the T4 buffer solution with 0.2 mM dGTP (matching base), 0.1 Unit T4 polymerase, and 0.05 μM hairpin DNA2. Base extension reactions were conducted in small PCR tubes at 37 °C for 1 h and then incubated at 70 °C for 20 min, prior to cool down and storage at 4 °C. For testing, small aliquots of the sample solutions were added to PDMS wells on the chip surfaces for PPi measurement on a Signatone probe station, as described in the measurement apparatus section. All samples were measured at room temperature (22 °C).
For in situ polymerase reactions, RCA DNA colonies were formed on the surface in the manner described previously with chelator deposited on the surface for PPi capture. For on-chip polymerase and control reactions, sample solutions were added to PDMS wells on chip surfaces and were incubated at 37 °C for 30 min in a high humidity enclosure. A number of control samples were characterized prior to proceeding with a series of polymerase reactions, including test solutions containing all base incorporation reaction ingredients before incubation at 37 °C, T4 buffer with PPi standards (25, 10, and/or 1 μM), T4 buffer only, buffer containing nucleotides but no polymerase, and T4 buffer with active or denatured polymerase among others as indicated. When present in the T4 buffer sample, nucleotides and T4 polymerase concentrations were 0.2 mM (each) and 0.1 U/μL, concentrations similar to those described for the off-chip reactions. All samples were measured at room temperature (22 °C) on a Signatone probe station.
Results and discussion
Surface characterization
A combination of surface analysis techniques (ellipsometry, atomic force microscopy (AFM), and fluorescence microscopy) was used to verify step-by-step monolayer chemical modification and functionality. Fluorescence microscopy results indicated similar functionalization efficiencies on oxidized silicon test substrates and the nanoplate sensors. An overview of the initial surface modification with the chelator only (no DNA) is illustrated in Scheme 1, Steps 1 to 3a. Piranha-cleaned SiO2 surfaces were functionalized with 1% aminopropyltriethoxysilane (APTES) in ethanol and then reacted with 2% glutaraldehyde (GA) in 1:1 ethanol:water. In our previous report we used an alternative modification technique with aldehyde functionalized silane (triethoxysilylbutyraldehyde, TESBA).5 While the number of steps increased due to the two step APTES + GA modification, we eliminated both the use of toluene and issues with multilayer deposition that resulted from wet toluene and polymerized TESBA. The GA-functionalized surfaces were exposed to amine-functionalized chelator in order to immobilized the chelator to the surface. The thickness of the functionalized silane layer (APTES with GA) was 14.2 ± 0.7 Å (n = 3) by ellipsometry (VASE), as expected. Chelator modified surfaces exhibited film thickness changes of ∼16 Å, as expected. In addition to ellipsometry, the expected mass of the chelator modification on silicon substrates has been detected by surface-sensitive TOF-SIMS.5
Expected pyrophosphate detection response
If based on net charge Q alone, the interaction of negatively charged PPi with the unmodified surface of a p-type accumulation mode device was expected to increase the number of positive carriers in the channel such that ΔQ > 0, increasing the drain current at a fixed potential and shifting the threshold voltage of the device more positive, according to ΔVt ∼ ΔQ/Ceff > 0, where Ceff is the effective capacitance.27,32 In Fig. 3, the I–V transfer characteristics of a p-type SOI-FET device with Vsd = 0.1V in accumulation mode were measured with different surface modifications (unmodified, chelator on silane and avidin-only) and different solution exposures. In acidic solution (pH 3 0.1M acetic acid), the current–voltage curve (in orange) was shifted to the left relative to pH 6–7 water (in purple) and pH 8 buffer (in pink), as expected. The curves in PBS buffer for the unmodified (dark pink and blue) and chelator + silane modified device (grey) were similar to each other. The unmodified device exposed to 25 μM PPi in 1x PBS pH8 buffer exhibited a shift to the right compared to the curve for deionized water, which was the expected direction for detection of negatively charged PPi based on surface charge effects on the p-type device. However the response for PPi was very similar to the control buffer of 1x PBS with Zn2+ containing no PPi on the unmodified device, indicative of screening of negatively charged PPi by Zn2+ and other cations in the buffer solution. In contrast, chelator-modified devices were highly responsive to a series of different PPi concentrations, even in 1x PBS with Zn2+ control buffer, as we describe in Figure S1 (ESI†).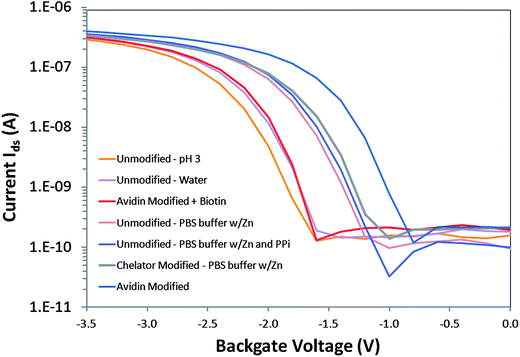 | ||
| Fig. 3 Current–voltage transfer characteristics of a p-type SOI-FET device with Vds = 0.1V in accumulation mode with different surface modifications (Unmodified, Chelator and silane modified and Avidin-only modified) and different solution exposures (pH 3 0.1M acetic acid, deionized water, pH 8 1x PBS buffer with Zn2+, pH 8 1x PBS buffer with Zn2+ plus 25 μM PPi, avidin in pH 8 borate buffer and avidin with biotin in pH 8 borate buffer). | ||
However, the interaction of PPi with the surface-immobilized chelator resulted in a sensor response that was in the opposite direction from that expected due to charge alone. It was more similar to that expected for positively charged ions interacting with a p-type device where ΔQ < 0 and ΔVt ∼ ΔQ/Ceff < 0. Upon exposure to PPi, we observed a decrease in drain current at a fixed potential and a shift in threshold voltage of the device to more negative values.5 In previously reported results we also describe exposure of chelator only-functionalized devices to solutions of different pH and observed no shift in threshold voltage,5 indicative of a primary sensing mechanism that appeared to be pH-independent and therefore should be subject to other parameters such as molecular or thin layer dipole effects on the field-effect transistor.
Another result opposite to that expected due to charge was also observed with the curve to the farthest right in Fig. 3 which was an unmodified device in pH 8 borate buffer with positively charged avidin homotetramer (∼4–5 nm diameter) electrostatically absorbed to the hydroxyl terminated SiO2 surface. Avidin is strongly positively charged with pI ∼ 10.5. Upon biotin exposure (red curve in Fig. 3), the curve previously observed for avidin shifts to the left, attenuating the change observed with avidin only, resulting in a current–voltage transfer characteristic more similar to the response for the unmodified device in water.Charge based response may be only one factor determining the direction of the threshold voltage upon surface binding events, in addition to molecular dipole moment and protein dipole/conformation effects.
One hypothesis explaining the observed threshold shift is that the dipole moment vector of the molecular assembly that includes the silane, glutaraldehyde and chelator complex changes dramatically upon PPi binding, similar to previous reports of uncharged species detected by field-effect devices.33 The molecular assembly can be individual molecules, domains of different, maybe even competing, molecules or the cumulative effect of a thin quasi-crystalline film. The chelator-zinc complex, once bound to the PPi, may influence the effective molecular dipole of the chelator such that the effective charge on the surface is positive, decreasing the drain current and producing a negative shift in the threshold voltage of the device, as observed in Figure S1.34
Using the GAMESS computational package, calculations of the dipole moment of the molecule consisting of the silane, glutaraldehyde, and chelator complex before and after PPi binding suggest that there is a dramatic reorientation of the dipole moment perpendicular to the device surface (see ESI). This structure-based dipole effect may account for the observation that the exposure of the functionalized surface to charge alone, in the form of zinc ions, does not result in a shift in the threshold voltage relative to control buffer without zinc ions. The charge distribution may not change the surface dipole unless the PPi binds to the chelator. For the DNA-based sensing applications discussed in this work, the cumulative effect of PPi binding to a chelator-modified device with DNA and avidin near the surface should be dominated by the PPi-to-chelator interaction. The avidin and DNA should be beyond the Debye length. Based on charge effects alone, the negatively charged PPi binding to the chelator near the surface should induce more positive carriers in the channel such that ΔQ > 0 and ΔVt ∼ ΔQ/Ceff > 0. However the more complicated, multi-component surface modification could change the properties of the chelator modification on the FET surface and introduce new dipole effects, water displacement or other effects not observed in the simple chelator modification.
Characterization of DNA colonies
For this work, both chelators and locally amplified DNA were attached on or near the SOI-FET sensor in order to detect PPi generated from enzymatic reactions on DNA immobilized on the device surface. The chelator and DNA co-deposition process is depicted in Scheme 1, steps 3b to 5. The aldehyde-terminated silane surface was functionalized with a 1:5 mixture of amine-chelator and amine-PEG12-COOH linker in order to add PPi-capture functionality and coupling sites for avidin amines.35 After co-deposition of chelator and PEG12 (1:5 molar ratio), the organic layer thickness was 35 Å, as reported in Table 1. This was a 21 Å difference from the previous layer reported in a previous section (APTES + GA, ∼14 Å) and similar to the film thickness observed from chelator-only modifications due to the relatively low density (submonolayer) of PEG linker sites on the surface. After amine groups on avidin were coupled to the carboxylic acid-terminated linkers by EDC coupling chemistry in pH 5 MES buffer,36 the substrate was incubated with biotinylated circle DNA.
In order to increase the number of PPi-generating sites on the surface without occupying surface area that could be utilized by the PPi chelator, we subjected immobilized DNA to rolling circle amplification (RCA) using the procedure depicted in Fig. 2a.37 After RCA, each DNA binding site contained many repeat units, each capable of undergoing individual base incorporation reactions. The DNA fragment copy number was dependent on the time elapsed during the RCA reaction.38 After an overnight reaction, the DNA colonies used in these experiments had approximately 50–100 copies of the same DNA fragment per colony. After hybridizing identical primers to the DNA at these 50–100 sites per colony, each primer was a potential polymerase reaction site (an average of 75 per colony) for a DNA base addition reaction.
Dye-labeling of the RCA DNA colonies was used to image their quantity and distribution on the surface after exposing the surface to different ratios of chelator and PEG linker. A Cy5 dye-labeled oligo (Cy5-CTGCAATGATACCGCGAGACCCA, 50nM in 1x TEST) complementary to one of the target DNA strands was hybridized to the amplified DNA on the surface at 50 °C for 30 to 60 min followed by washing with 1x TEST buffer before fluorescence imaging. Compared to the 1:1 surface, the 1:5 and 1:9 surfaces in Fig. 2c and 2d indicate increased coverage of fluorescently labeled DNA, corresponding to more DNA as the PEG linker concentration increased. Ellipsometry data for 1:1 and 1:5 surfaces in Table 1 support the same trend, with a greater average organic layer thickness measured for the 1:5 chelator:PEG linker surfaces, as expected. As shown in Fig. 2e, we typically used the 1:5 mixture for preparation of FET surfaces in order to avoiding the clustering that we observed with 1:9 samples. The surface coverage with the 1:1 mixture was not used because the DNA colony distribution was sparse and the likelihood of DNA colony attachment near the active sensor area was too low.
In addition to fluorescence microscopy, RCA DNA modified surfaces were characterized by AFM to complement larger area fluorescence microscopy images and to directly assess DNA colony size by height. In these studies, avidin was electrostatically bound to freshly piranha cleaned SiO2 on Si substrates. The presence of avidin was confirmed by a number of dye-labeling techniques, particularly fluorescence labeling of biotinylated DNA attached to avidin or Cy5-dU incorporation on immobilized DNA. As described in previous sections biotinylated circle DNA can be attached to avidin and then sequences can be amplified by RCA. Fig. 4a is a high contrast fluorescence microscopy image of Cy5-dU labeled RCA DNA on avidin with regions of high DNA colony coverage (on left) and regions with relatively few DNA colonies (right). In order to create these two distinct regions, we used a plastic pipette tip to lightly scrape some of the avidin from the surface prior to DNA attachment and this interface is marked by a dotted line in Fig. 4a and can also be observed in the topographical AFM images in Fig. 4b and 4c. From the image in Fig. 4a we can also observe that the non-specific binding of the DNA circles and primers on the surface is minimal (low fluorescence background observed), which is consistent with ellipsometry and AFM results and measurements on the devices. In our experiments, non-specific binding of avidin is more likely than non-specific DNA interactions as we have observed with some fluorescence, ellipsometry and AFM data. However we can control non-specific avidin binding with optimized rinsing procedures (buffer composition, flushing volume and number of times) and choosing compatible materials that we can selectively coat with protein-resistant molecules, such as PEG-based blocker molecules.39 In parallel work, we have verified the chemical and thermal stability of the DNA colony units as well as their attachment to the surface. We have evaluted these areas (avidin rinsing or removal by higher temperature incubations and DNA colony stability) with the same characterization techniques we have previously described, such as fluorescence, ellipsometry and AFM. These are areas of process and materials development that we continually optimize.
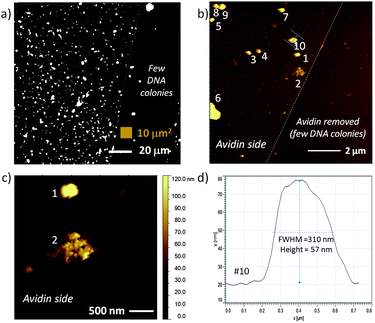 | ||
| Fig. 4 Fluorescence image and AFM topography of avidin and RCA DNA colonies on a flat SiO2 on Si test substrate: a) fluorescence image of Cy5-dU labeled DNA colonies with a region on the right with few DNA colonies; b) 100 μm2 area equivalent to small brown box in (a) with numbered clusters on the left side identified for cross-section analysis, c) region that contains two large features – one with typical DNA colony profile and second with very different topography indicative of a combined DNA colony, and d) cross-section of feature #10, an RCA DNA colony with height ∼57 nm and full width at half the maximum height (FWHM) of 310 nm. | ||
The smallest DNA colony features measured in Fig. 4 (possibly single DNA colonies) were 42 ± 9 nm (n = 4) height with width of 196 ± 21 nm. While the AFM height is known to be accurate to within 0.1 nm on most AFM instruments, the accuracy of the width measurement is typically limited by the width of the AFM tip, where the tip itself can be traced by the smaller diameter surface protrusion (pseudo-tip). Including the larger DNA features numbered in Fig. 4b (which may be more than one colony), the average feature height was 67 ± 39 nm (n = 9) with width of 262 ± 91 nm (note the larger standard deviation). These values are within the expected range, with a DNA colony size of 100 to 300 nm expected in buffer.
Detection of PPi from T4 polymerase reactions in solution
In order to demonstrate PPi sensing by SOI-FET devices modified with both chelator and DNA, we first exposed the devices to 25 μM PPi standard solution, as shown in Fig. 5a and 5b, and observed a +0.41 V and +0.42 V shift in threshold voltage in the positive direction. These values were obtained from two similar devices on the same chip. The results observed for these devices (ΔVt > 0) were consistent with the initially expected shift dictated by charge effects induced by the negatively charged PPi on the p-type device (ΔQ > 0) but opposite the effect observed in the PPi titration experiments reported in Figure S1. Since the surface modification contains avidin, DNA and a linker molecule in addition to chelator, there may be complicated interactions on the surface and between different domains on the surface (disordered monolayer) that are not observed in the chelator only modified devices.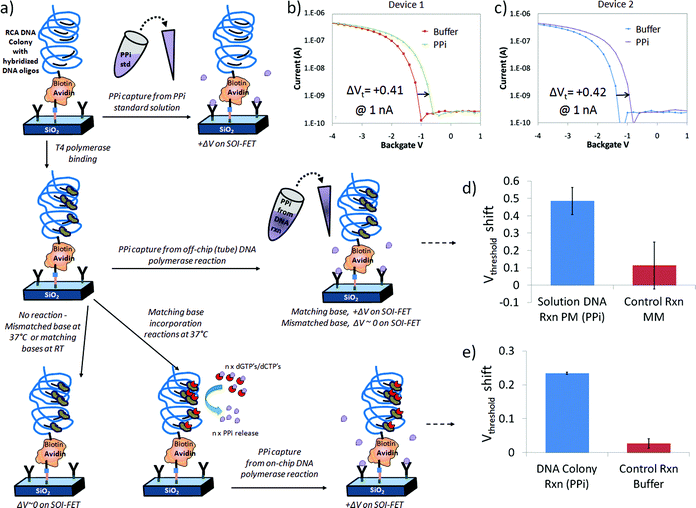 | ||
| Fig. 5 Label-free detection of PPi on chelator- and DNA-modified FET devices: a) schematic of a simplified chelator + DNA colony device surface as it is exposed to three different PPi sources including PPi standard solutions (top row), PPi generated in a reaction tube by polymerase reactions incorporating a matching DNA on linear DNA in solution (middle row), and PPi generated by multi-base (dGTP + dCTP) incorporation reactions on surface immobilized RCA DNA colonies (bottom row), b–c) response of two different devices on same chip to 25 μM PPi standard solution in 1x PBS buffer, d) bar graph representing average of 3 measurements on same device after exposure to perfect match (PM) dGTP off-chip reaction solution, resulting in a positive change in threshold voltage (+0.49 V) compared to the control mismatch nucleotide solution (+0.11 V) and e) bar graph representing the average response on same device after 3 independent on-chip DNA polymerase reactions at 37 °C, generating PPi (+0.23 V), compared to incubations in reaction buffer at 22 °C with no DNA polymerase reactions (+0.03 V). | ||
Following this initial test, we progressed to detection of PPi from off-chip DNA reactions with T4 polymerase in the presence of linear DNA and dGTP perfect match (PM) or dTTP mismatch (MM) nucleotide triphosphates, as shown in Fig. 5c and 5d. The current–voltage transfer characteristic was initially measured in Tris control buffer. PPi sensing from DNA base incorporation reactions in solution was observed on several devices but for simplicity in this discusion we focus on the results from one device as a function of a series of independent experiments. When the FET sensor was exposed to an off-chip reaction mixture containing hairpin DNA2 (sequence information provided in the experimental section) and the matching base (dGTP) with T4 polymerase, we detected a positive change in threshold voltage of +0.49 ± 0.08 V (n = 3) for the dGTP perfect match solutions at 1nA relative to the control buffer (n = 3, normalized to ΔVt = 0) as presented in Fig. 5d. The shift in threshold voltage was in the same direction as the devices exposed to 25 μM PPi standard solution in Fig. 5a and 5b. Thus, the device response was consistent with the detection of PPi generated from T4 + dGTP base incorporation reactions on hairpin DNA2 in a reaction tube, as expected. After a 0.1M acetic acid wash to remove the PPi bound to the Zn2+ and chelator complex, the chip was exposed to a control solution containing hairpin DNA2, polymerase and a non-matching base (dTTP), which resulted in a threshold voltage shift of +0.11 ± 0.14 V (n = 3) compared to the initial buffer condition.
Detection of PPi from DNA colony reactions on surface
In the final set of experiments, illustrated in Fig. 5a and 5e, a T4 polymerase enzymatic reaction solution containing more than one base (dGTP + dCTP) was added to the chelator and DNA colony modified chip and measured before and after incubation at 37 °C. Reaction times for these on-chip experiments were typically 30 min in order to closely follow the typical assay that we performed for the hairpin DNA. Control experiments with buffer solution and control solutions missing the full complement of base incorporation reaction components did not typically induce changes in threshold voltage, as expected. However, PPi generated from a positive T4 polymerase reaction on DNA incorporating the matching bases dGTP and dCTP produced a threshold voltage shift to more positive potentials, as observed in Fig. 5e. As in previously described experiments, the PPi could be stripped from the chelators with an 0.1M acetic acid rinse and the response in buffer was similar to that observed before the base incorporation reaction.For the bar graph in Fig. 5e, where PPi was generated from T4 polymerase reactions on DNA colonies immobilized on chip after exposure to a multi-base (dGTP and dCTP) extension solution and incubation at 37 °C, the observed threshold voltage shift was +0.23(4) ± 0.00(3) V from one device and n = 3 independent solution exposures or experiments compared to initial buffer measurements. In comparison, for three separate control reactions missing T4 polymerase, but still containing other components and the nucleotide bases dGTP and dCTP an I–V transfer characteristic similar to the reaction buffer, with a minimal threshold voltage shift of +0.03 ± 0.01 V was observed, as expected. Therefore the shift in threshold voltage observed in Fig. 5d after DNA polymerase reactions on DNA colonies on-chip was in the same direction as the devices exposed to 25 μM PPi standard solution in Fig. 5a and 5b and the hairpin DNA 2 PM solution in Fig. 5c and is consistent with the label-free chelator-based detection of PPi generated on the SOI-FET surface.
The magnitude of the shift was approximately half that of the other experiments with 47% of the hairpin DNA2 shift and 55% of the 25 uM PPi standard threshold voltage shift, which was better than expected. Using reaction times similar to what was used for tube reactions (∼30 min) we were able to generate PPi in sufficient concentration to bind to the surface immobilized chelators on the SOI-FET sensor and detect the response. We did not find it necessary to modify our polymerase reaction protocol significantly in transitioning between a tube-based assay and a surface-based assay, which may be an advantage of the separate functions of the PPi-generating DNA colony and PPi-capturing chelator. The chelator-based sensing mechanism does not require high-density DNA loading on the surface, which can affect how well polymerase or other enzymes can interact with the DNA.
With an average of 75 PPi-generating reactions per DNA colony, as described in a previous section, at least 2500 PPi molecules were produced per colony for a single base incorporation reaction. Given the observed surface coverage of ∼1 colony/μm2 for the 1:5 chelator to linker deposition condition reported in Fig. 2d, a 2 mm2 reaction area on the chip surface and a solution volume of 2 μL, this reaction generated at least 7.5 × 109PPi molecules, corresponding to a PPi concentration of 6.2 nM if all the PPi molecules dispersed into solution before being captured on the surface. It is possible that the fluorescence labeling method used to estimate ∼1 colony/μm2 underestimated the number of DNA circles or colonies undergoing polymerase reactions and generating PPi. With the T4 polymerase reaction, the concentration of PPi molecules increases with time given a sufficient amount of DNA and nucleotides, so the concentration of PPi in solution for a multi-base (dGTP and dCTP) extension reaction was expected to be much higher than 6.2 nM after 30 min. We expected the DNA colony-based reactions to produce at least 1–10 μM PPi based on SOI-FET results obtained on tube-based T4 polymerase assays, separate DNA colony measurements on other types of electrical devices, and luminescence based assays. While we did not conduct a full titration experiment for the chelator and DNA colony modified chips, as we did for the chelator-only modified FET devices described in Figure S1, we did test the response of chelator + DNA devices to 1 μM and 10 μM PPi standards in T4 polymerase reaction buffers and did observe a positive shift in threshold voltage with increasing PPi concentration, with the size of shift consistent with the results reported in Fig. 5.
Conclusions
We describe surface-sensitive electrical detection using a molecular interface on silicon-on-insulator (SOI) FET devices designed to capture diffusible pyrophosphate (PPi) generated by polymerase reactions on surface-immobilized DNA. We demonstrated three types of PPi detection by chelator-modified FET: PPi standards in solution, PPi generated from enzymatic DNA reactions on linear DNA in solution, and PPi generated from enzymatic DNA reactions on surface-bound DNA. This work is unique and important in several aspects. The PPi-specific chelator that we have synthesized is used for the first time for label-free (non-fluorescent, non-luminescent) electrical detection of enzymatic DNA base incorporation reactions, demonstrating a key function for potential DNA analysis applications. The response is concentration dependent and reversible. These results are enabled by a specific set of well-characterized surface modifications that enable the co-attachment of chelator on the surface in close proximity to surface-bound DNA reaction sites. In order to minimize the occupation sites of DNA compared to the chelator capture molecules on the sensor surface, we employ an amplification procedure (RCA) that generates a large number of PPi signaling molecules in the vicinity of one DNA attachment site. Coupled with the chelator-modified field-effect sensor, this represents a relatively new approach to electrical detection of enzymatic DNA base incorporation reactions and is scalable within the limits of the sensitivity of the sensor. Although the results described in this work were from PCR amplified DNA, we have also demonstrated RCA DNA colony formation from total genomic DNA on patterned silicon oxide substrates in parallel studies. Therefore, the RCA and SOI-FET detection methods described in this paper can be extended to complex DNA samples from a variety of sources without pre-amplification or selection.With these results a label-free approach for sensing DNA polymerase reactions on surface-immobilized DNA using PPi-sensitive sensors can be optimized for a variety of applications. This work can be applied to the development of massively parallel electronic sensor arrays for PPi-sensing based applications such as portable, integrated systems for DNA analyses, such as genotyping or DNA sequencing.40,41 In addition, the general principle of chelator-modification of scalable SOI-FET devices can be extended to highly parallel electronic sensing of other biochemical reactions for a wide variety of applications beyond DNA sequencing.
Acknowledgements
We thank Jingwu Zhang, Hsiao C. Lim, Mun Hua Tan, Mineo Yamakawa, Ryan Murray, Esmeraldo Gorecho, Misbah Palla, and Liming Wang for assistance with sample preparation and sample analysis methods development, Scott Lancaster (NMML, Intel New Mexico) for high resolution SEM images, the staff at Birck Nanotechnology Laboratory at Purdue University and the Micro and Nanotechnology Laboratory at UIUC for FET processing, and Stanford Nanofabrication Facility (NNIN, NSF Grant ECS-9731293) for providing instrument access for ellipsometry. Bobby Reddy, Jr. and Brian Ross Dorvel were supported by a grant from NIH R01-CA20003 at UIUC.References
- J. K. Heinonen, Biological role of inorganic pyrophosphate, Kluwer Academic Publishers, London, U.K., 2001 Search PubMed.
- S. K. Kim, D. H. Lee, J. I. Hong and J. Yoon, Acc. Chem. Res., 2009, 42, 23–31 CrossRef CAS.
- M. Ronaghi, S. Karamohamed, B. Pettersson, M. Uhlen and P. Nyren, Science, 2008, 281, 363–365 CrossRef.
- M. Ronaghi, M. Uhlén and P. Nyrén, Science, 1998, 281, 363–365 CrossRef CAS.
- D. J. Liu, G. M. Credo, K. Wu, H. C. Lim, X. Su, O. Elibol, R. Bashir and M. Varma, Chem. Commun., 2011, 47, 8310–8312 RSC.
- A. Hierlemann and R. Gutierrez-Osuna, Chem. Rev., 2008, 108, 563–613 CrossRef CAS.
- J. Wang, Biosens. Bioelectron., 2006, 21, 1887–1892 CrossRef CAS.
- Y. Maruyama, S. Terao and K. Sawada, Biosens. Bioelectron., 2009, 24, 3108–3112 CrossRef CAS.
- S. Roy, X. J. Chen, M. H. Li, Y. F. Peng, F. Anariba and Z. Q. Gao, J. Am. Chem. Soc., 2009, 131, 12211–12217 CrossRef CAS.
- J. H. Chua, R.-E. Chee, A. Agarwal, S. M. Wong and G.-J. Zhang, Anal. Chem., 2009, 81, 6266–6271 CrossRef CAS.
- J. M. Rothberg, W. Hinz, T. M. Rearick, J. Schultz, W. Mileski, M. Davey, J. H. Leamon, K. Johnson, M. J. Milgrew, M. Edwards, J. Hoon, J. F. Simons, D. Marran, J. W. Myers, J. F. Davidson, A. Branting, J. R. Nobile, B. P. Puc, D. Light, T. A. Clark, M. Huber, J. T. Branciforte, I. B. Stoner, S. E. Cawley, M. Lyons, Y. Fu, N. Homer, M. Sedova, X. Miao, B. Reed, J. Sabina, E. Feierstein, M. Schorn, M. Alanjary, E. Dimalanta, D. Dressman, R. Kasinskas, T. Sokolsky, J. A. Fidanza, E. Namsaraev, K. J. McKernan, A. Williams, G. T. Roth and J. Bustillo, Nature, 2011, 475, 348–352 CrossRef CAS.
- A. Poghossian, M. H. Abouzar, F. Amberger, D. Mayer, Y. Han, S. Ingebrandt, A. Offenhausser and M. J. Schoning, Biosens. Bioelectron., 2007, 22, 2100–2107 CrossRef CAS.
- S. J. Osterfeld, H. Yu, R. S. Gaster, S. Caramuta, L. Xu, S. J. Han, D. A. Hall, R. J. Wilson, S. H. Sun, R. L. White, R. W. Davis, N. Pourmand and S. X. Wang, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 20637–20640 CrossRef CAS.
- Y. F. Wu, S. Q. Liu and L. He, Anal. Chem., 2009, 81, 7015–7021 CrossRef CAS.
- F. Ricci, N. Zari, F. Caprio, S. Recine, A. Amine, D. Moscone, G. Palleschi and K. W. Plaxco, Bioelectrochemistry, 2009, 76, 208–213 CrossRef CAS.
- S. Taylor, S. Smith, B. Windle and A. Guiseppi-Elie, Nucleic Acids Res., 2003, 31 Search PubMed.
- Y. Han, A. Offenhausser and S. Ingebrandt, Surf. Interface Anal., 2006, 38, 176–181 CrossRef CAS.
- T. Uno, T. Ohtake, H. Tabata and T. Kawai, Jpn. J. Appl. Phys., 2004, 43, L1584–L1587 CrossRef CAS.
- S. Ingebrandt, Y. Han, F. Nakamura, A. Poghossian, M. J. Schoning and A. Offenhausser, Biosens. Bioelectron., 2007, 22, 2834–2840 CrossRef CAS.
- A. Poghossian, A. Cherstvy, S. Ingebrandt, A. Offenhausser and M. J. Schoning, Sens. Actuators, B, 2005, 111, 470–480 CrossRef.
- T. He, D. A. Corley, M. Lu, N. H. Di Spigna, J. L. He, D. P. Nackashi, P. D. Franzon and J. M. Tour, J. Am. Chem. Soc., 2009, 131, 10023–10030 CrossRef CAS.
- P. R. Nair and M. A. Alam, IEEE Trans. Electron Devices, 2007, 54, 3400–3408 CrossRef CAS.
- C. W. Fuller, L. R. Middendorf, S. A. Benner, G. M. Church, T. Harris, X. H. Huang, S. B. Jovanovich, J. R. Nelson, J. A. Schloss, D. C. Schwartz and D. V. Vezenov, Nat. Biotechnol., 2009, 27, 1013–1023 CrossRef CAS.
- J. Guo, L. Yu, N. J. Turro and J. Y. Ju, Acc. Chem. Res., 2010, 43, 551–563 CrossRef CAS.
- B. Schweitzer, S. Wiltshire, J. Lambert, S. O'Malley, K. Kukanskis, Z. Zhu, S. F. Kingsmore, P. M. Lizardi and D. C. Ward, Proc. Natl. Acad. Sci., 2000, 97, 10113–10119 CrossRef CAS.
- H. Nakano, Nucleic Acids Res., 2005, 33, e150 CrossRef.
- O. Knopfmacher, A. Tarasov, W. Y. Fu, M. Wipf, B. Niesen, M. Calame and C. Schonenberger, Nano Lett., 2010, 10, 2268–2274 CrossRef CAS.
- O. H. Elibol, J. Bobby Reddy and R. Bashir, Appl. Phys. Lett., 2008, 92, 193904 CrossRef.
- B. Reddy, B. R. Dorvel, J. Go, P. R. Nair, O. H. Elibol, G. M. Credo, J. S. Daniels, E. K. C. Chow, X. Su, M. Varma, M. A. Alam and R. Bashir, Biomed. Microdevices, 2011, 13, 335–344 CrossRef CAS.
- R. Tero, N. Misawa and H. Watanabe, e-J. Surf. Sci. Nanotechnol., 2005, 3(237), 237–243 CrossRef CAS.
- A. Ulman, An Introduction to Ultrathin Organic Films, Academic Press, San Diego, 1991 Search PubMed.
- B. R. Takulapalli, ACS Nano, 2010, 4, 999–1011 CrossRef CAS.
- I. Goykhman, N. Korbakov, C. Bartic, G. Borghs, M. Spira, J. Shappir and S. Yitzchaik, J. Am. Chem. Soc., 2009, 131, 4788–4794 CrossRef CAS.
- D. Alloway, M. Hofmann, D. Smith, N. Gruhn, A. Graham, R. Colorado, V. Wysocki, R. Lee, P. Lee and N. Armstrong, J. Phys. Chem. B, 2003, 107, 11690–11699 CrossRef CAS.
- H. Otsuka, Y. Nagasaki and K. Kataoka, Langmuir, 2004, 20, 11285–11287 CrossRef CAS.
- S. W. Metzger, M. Natesan, C. Yanavich, J. Schneider and G. U. Lee, J. Vac. Sci. Technol., A, 1999, 17, 2623–2628 CAS.
- J. Hu and C.-y. Zhang, Anal. Chem., 2010, 82, 8991–8997 CrossRef CAS.
- M. Hori, H. Fukano and Y. Suzuki, Biochem. Biophys. Res. Commun., 2007, 352, 323–328 CrossRef CAS.
- M. Palma, J. J. Abramson, A. A. Gorodetsky, E. Penzo, R. L. Gonzalez, M. P. Sheetz, C. Nuckolls, J. Hone and S. J. Wind, J. Am. Chem. Soc., 2011, 133, 7656–7659 CrossRef CAS.
- K. V. Voelkerding, S. A. Dames and J. D. Durtschi, Clin. Chem., 2009, 55, 641–658 CAS.
- E. R. Mardis, Trends Genet., 2008, 24, 133–141 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional SOI-FET data from chemically modified devices, 3D renderings of the chelator molecule and pKa information. See DOI: 10.1039/c2an15930a |
| This journal is © The Royal Society of Chemistry 2012 |