Femtosecond lasers for mass spectrometry: Proposed application to catalytic hydrogenation of butadiene
Orla
Kelly
,
Martin J.
Duffy
,
Raymond B.
King
,
Louise
Belshaw
,
Ian D.
Williams
,
Jacinto
Sá†
,
Chris R.
Calvert
and
Jason B.
Greenwood
*
Centre for Plasma Physics, School of Maths and Physics, Queen's University Belfast, Belfast, BT7 1NN, UK. E-mail: j.greenwood@qub.ac.uk
First published on 9th November 2011
Abstract
Mass spectra from the interaction of intense, femtosecond laser pulses with 1,3-butadiene, 1-butene, and n-butane have been obtained. The proportion of the fragment ions produced as a function of intensity, pulse length, and wavelength was investigated. Potential mass spectrometry applications, for example in the analysis of catalytic reaction products, are discussed.
Introduction
Of all possible chemical analysis techniques, mass spectrometry is the one which typically provides the highest sensitivity, an attribute which strongly depends on the nature of the ionisation source used. For molecular species, there is also a need for the ionisation method to produce parent molecular ions or characteristic fragment ions when there is a complex mixture of products. This becomes more important with increasing molecular size as different parent molecules can yield fragment ions with the same molecular mass.The standard method for gas phase ionisation is by electron impact since it is simple, cheap and well characterised for most molecules. An electron collision energy of 70 eV is usually employed as this provides the maximum energy transfer to the molecule, giving the most efficient ionisation. However, for complex molecules this can result in a plethora of fragments in the mass spectrum, with negligible production of parent ions. While this can yield structural information, for identification of the molecular species present a spectrum dominated by the parent molecular ion is far more desirable. The ionisation efficiency is also typically restricted to a maximum of 0.1% due to the electron beam current density being limited by space charge.
Recently single photon ionisation has been gaining recognition as an alternative method since the energy deposition to the molecule during ionisation can be reduced by operating much closer to threshold. Although photoionisation cross sections tend to be two orders of magnitude lower than for electron impact at 70 eV, a much greater fraction of the ions produced are due to the intact parent cation. Until recently, vacuum ultraviolet sources lacked the brightness to provide a sufficiently high ionisation efficiency but the use of electron beam excimer lasers (e.g.Ar2*, 126 nm, 9.9 eV), the 9th harmonic of a Nd:YAG laser (118 nm, 10.5 eV), and F2 lasers (157 nm, 7.9 eV) have been used to identify organic molecules in chemical samples in the last few years.1 Ideally the wavelength is tuned to just above the ionisation potential of the species of interest. To date this type of work has been limited to studies at synchrotrons which allow complex mixtures to be studied, such as those found in combustion.2 However, the wider applicability of such sources to mass spectrometry is naturally limited to a few locations in the world.
As the photon energies of most conventional lasers are less than molecular ionisation potentials,1 application of photoionisation to mass spectrometry is usually implemented via multiphoton ionisation. Unless there is a strong resonant enhancement from excited states, this necessitates the use of high intensity short pulse lasers. With nanosecond lasers, ionisation proceeds by a ladder switching process whereby population of excited levels results in dissociation or transfer to other states, before further photons are absorbed to liberate an electron. By contrast, femtosecond laser ionisation is more direct, proceeding via ladder climbing through virtual states with any dissociation usually following the ionisation. Comparison between these two techniques shows that femtosecond lasers have much higher ionisation efficiencies and produce less fragmentation.3
For situations where isomers need to be identified, femtosecond lasers can also produce more distinct differences in mass spectra than electron impact. This has recently been demonstrated4 for 1-butene and cis-2-butene with 175 fs pulses at a wavelength of 800 nm, while even greater contrast can be obtained by optimally shaping the intensity envelope of the femtosecond pulse.5
Femtosecond lasers have yet to be widely employed in mass spectrometry due to the cost and expertise required. However, in the last few years many commercial femtosecond laser systems have come onto the market providing ‘turnkey’ operation at reduced cost. In this paper, we demonstrate that under the appropriate conditions, femtosecond lasers are capable of ionising hydrocarbon molecules without generating the substantial dissociation typically observed by electron impact. We concentrate on the molecules 1,3-butadiene C4H6, 1-butene C4H8, and n-butane C4H10, due to interest in the catalytic hydrogenation of 1,3-butadiene over Pd/Al2O36catalysts and more recently over Au modified metallic organic frameworks (MOFs).7Isooctane, a high octane gasoline component, can be produced by dimerisation of isobutene, which is currently produced from catalytic cracking of gas oil. However the isobutene stream has traces of butadiene. The presence of traces of 1,3-butadiene (up to 1%) in the isobutene can lead to poisoning of the dimerisation catalyst, necessitating its removal.
An apparatus is currently under construction to investigate this and other catalytic processes using a femtosecond laser coupled to a time of flight mass spectrometer to obtain time-resolved mass spectra of the catalytic reaction products. This paper demonstrates that this instrument needs a femtosecond laser for ionisation because an electron beam cannot be relied upon to distinguish between the products of butadiene catalysis.
Experimental method
The experimental apparatus in Belfast used for the current study consists of a novel electrostatic ion trap within which molecular and fragment ions are generated by a focused femtosecond laser pulse. It consists of a series of apertured plates which generate the potential surface shown in Fig. 1. Ions can oscillate along stable linear trajectories between two mirrors in a manner analogous to a laser cavity. As the device is electrostatic, the trapping conditions are mass independent but the oscillation periods are proportional to the square root of the mass. By acquiring the image charge on several pickup rings located near the centre of the device, frequency analysis of this signal allows high resolution mass spectra to be obtained over an unlimited mass range. Further details of the apparatus and analysis methods can be found in a recent publication.9 | ||
| Fig. 1 Photograph of the electrostatic ion trap and the typical potential energy surface8 produced by the apertured electrodes, with decreasing potentials on electrodes V7 − V1 forming two electrostatic mirrors. The trap has cylindrical symmetry and ions are generated between electrodes V7 and V6 in one of the mirror regions by a femtosecond laser as shown. Ion oscillations within the trap are confined axially by the electrostatic mirrors and radially by the einzel lens. High-resolution mass spectra are extracted via CHIMERA analysis of the image charge detection signal. | ||
Femtosecond pulses at 800 nm were produced by a Coherent LIBRA Ti:Sapphire regenerative amplifier to give bandwidth limited pulses of 100 fs duration with an energy of nearly 1 mJ. Femtosecond pulses at 1400 nm with pulse energies of 60 μJ were generated by optical parametric amplification of the 800 nm pulses using a Coherent OPerA module. A 15 cm focal length spherical lens brought the laser pulses to a focus between the two plates V7 and V6 in one of the electrostatic mirrors, as shown in Fig. 1. The pulses were linearly polarised in a direction along the trap axis.
The intensity was varied by attenuating the pulse energy using a half wave plate in combination with a linear polarizer. The pulse intensities were estimated by introducing xenon gas and comparing the yields of single, doubly and triply charged ions as a function of intensity to previous results using similar pulses.10,11 Throughout the paper, pulse intensities are stated corresponding to the spatial and temporal peak of the pulse, and these have a 50% uncertainty associated with them.
For each experiment, the specific gas under investigation was leaked into the vacuum chamber (with a base pressure of 10−9 mbar), until a pressure of around 10−7 mbar was attained. Once generated, the ions were accelerated by the electric field of the mirror into the drift region, where the ion bunch charge was imaged on three cylindrical electrodes. The charge pulse was converted to a voltage by a cold field-effect transistor and charge sensitive pre-amplifier (Amptek CoolFET®) and recorded as a function of time. Radial confinement of ions was attained by applying an appropriate potential to the lens electrodes.
Fig. 2a shows signal obtained on the pickup ring at the geometric centre of the trap from the interaction of 100 fs, 800 nm pulses with 1,3-butadiene at a peak intensity of 4.0 × 1014 Wcm−2. A series of periodic impulses occur due to oscillating butadiene radical cations. Smaller contributions from fragment ions can also be observed as a series of peaks with higher oscillation frequencies. From an absolute calibration of the pickup ring,12 it can be determined that about 104 ions are initially generated by each laser pulse. In the first few oscillations the peak reduces in magnitude due to loss of ions which are on unstable trajectories. The subsequent trap lifetime of the ions (about 10 ms) is then determined predominantly by losses due to collisions with background gas.
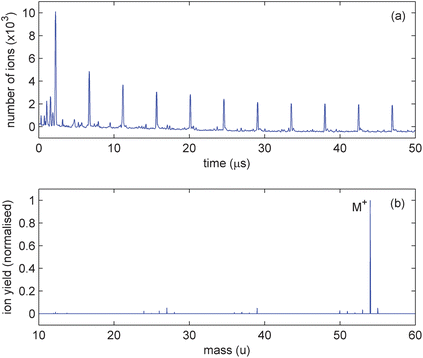 | ||
| Fig. 2 Results of the irradiation of 1,3-butadiene by 800 nm, 100 fs pulses, at an intensity of 4.0 × 1014 Wcm−2; (a) central pickup ring image charge acquisition for the first 50 μs; (b) mass spectrum obtained from frequency analysis of data in (a). | ||
Ion oscillation frequencies can be extracted using a standard Fast Fourier Transform algorithm but as the signal is non-sinusoidal, each ion generates not only a fundamental frequency but also many harmonics. This makes generation of a mass spectrum difficult when many different ions are present as there is no one-to-one correspondence between frequency and mass. We have radically improved this situation with the development of an alternative frequency analysis algorithm, CHIMERA,9 which is based on the use of a comb function as a basis rather than sinusoids. Using data from two pickup rings, a full mass spectrum can be generated with higher resolution than that achievable via a simple Fourier transform. CHIMERA analysis of the data in Fig. 2a yields the mass spectrum shown in Fig. 2b. Only the first 1 ms of the data is analysed to ensure that the relative ion yields are not influenced by differences in trapping lifetimes.
To compare the relative yield of parent to fragment ions the efficiency with which different ions are detected and trapped must be considered. The pickup rings, unlike microchannel plate detectors, are sensitive only to the ion charge and do not depend on mass or velocity, so no correction for detector efficiency is necessary (except to account for the additional charge of doubly charged ions). As the molecular gas is at room temperature, the initial velocity of the parent ions is essentially zero. However, fragment ions can have significant initial velocities as they gain energy in the dissociation process, especially for low mass fragments or multiply charged ions resulting from Coulomb explosion. The acceptance solid angle of the trap is dependent on the potential energy surface employed and the dissociation energy of the fragment ions. For the potentials used in these experiments, ion optics simulations8 indicate that for ions generated on the trap axis, 100% are trapped on stable trajectories if they have initial kinetic energies of less than 1 eV. In a separate experiment using time of flight analysis with a narrow acceptance solid angle, we were able to measure the kinetic energy release of fragment ions. For all but the lightest ions, e.g.protons, and multiply charged atomic ions which had low total yields, the initial kinetic energies were less than 1 eV. Therefore, our mass spectra provide a reliable measurement of the relative ion yields.
Results
800nm
In Table 1, data on fragment ion appearance energies obtained from previous studies are shown for the three molecules. Fig. 3 and Fig. 4 show mass spectra obtained for 1-butene and n-butane respectively at three different intensities at a wavelength of 800 nm.| 1st Ionisation Energy (eV) | 1,3-butadiene | 1-butene | n-butane |
|---|---|---|---|
| 9.07 | 9.55 | 10.53 | |
| Appearance Energy (eV) | |||
| –H loss | 11.4 | 11.3 | 11.7 |
| −CH3 | 11.3 | 11.8 | 11.2 |
| −CH4 | 11.2 | ||
| −CH5 | 12.4 | 13.8 | 13.4 |
| −C2H4 | 15.4 | 11.7 | |
| −C2H5 | 13.6 | 12.6 | |
| −C2H6 | 11.7 | ||
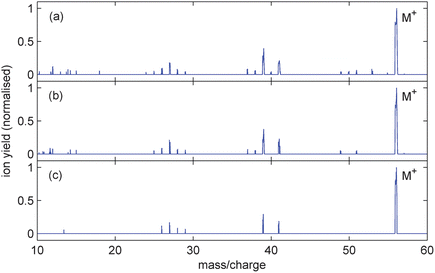 | ||
| Fig. 3 Mass spectra from 1-butene (C4H8) ionised with 800 nm, 100 fs pulses of peak intensity (a) 4.0 × 1014 Wcm−2, (b) 1.0 × 1014 Wcm−2 and (c) 6.6 × 1013 Wcm−2. The corresponding parent ion yields are 39%, 43% and 51% respectively. | ||
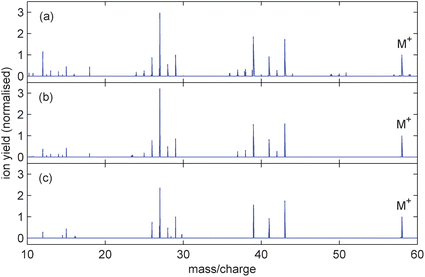 | ||
| Fig. 4 Mass spectra from n-butane (C4H10) ionised with 800 nm, 100 fs pulses of peak intensity (a) 4.0 × 1014 Wcm−2, (b) 1.0 × 1014 Wcm−2 and (c) 6.6 × 1013 Wcm−2. The corresponding parent ion yields are 8.1%, 8.9% and 9.3% respectively. | ||
While it is evident from Fig. 2b that intact butadiene cations dominate, parent ion production is not as strong for butene and considerably weaker for butane. As the valence electrons along the molecular backbone become more localised with increasing hydrogenation, leading to fewer double bonds, the ionisation potential increases (see Table 1). Meanwhile, the appearance energies of various fragment ions in each molecule, associated with loss of the same neutral groups, remain relatively unchanged. For butane this means that the ionisation potential is energetically much closer to possible dissociation channels.
In Fig. 5(a,c,e) the total ion yields are plotted as a function of intensity for the three molecules. Each graph shows a strong intensity dependence at low intensities with a more modest increase in yield at higher intensities. This behaviour is characteristic of the peak intensity reaching a saturation value for which the ionisation probability approaches 100% at the centre of the focus. Above this value, the yield increases with intensity to the power of 1.5 due to expansion of iso-intensity surfaces. For butadiene, butene and butane this saturation intensity is about 1, 1.5, and 2 × 1014 Wcm−2 respectively. While these values are slightly higher than what might be expected for the ionisation potentials of these molecules, it has been previously noted that induced polarisation of the molecule and molecular orbital configurations can increase saturation intensities for molecules compared to atoms with a similar ionisation potential.14,15
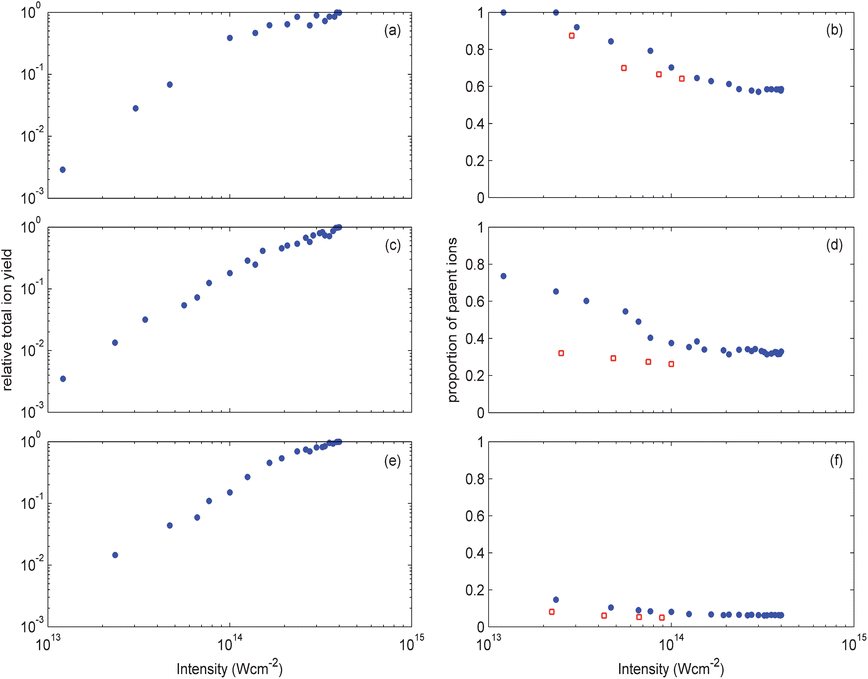 | ||
| Fig. 5 Total ion yields (left) and the fraction which are parent ions (right) for (a,b) butadiene, (c,d) butene and (e,f) butane as a function of pulse peak intensity. Blue circles correspond to results obtained with pulses of 100 fs duration while the red squares are data from longer pulses of: (a,b) 350 fs; (c,d) 400 fs; (e,f) 450 fs. | ||
An intensity of 1014 Wcm−2 corresponds to a Keldysh parameter16 of slightly less than 1 for these molecules, indicating that tunnelling or over-the-barrier ionisation is the most significant ionisation mechanism. However, most of the ions are generated from lower intensity regions of the focussed pulse for which the volume of interaction with the gas molecules is considerably greater. Therefore, we expect that multi-photon ionisation is generating most of the ions we observe, even at the highest intensities.
In Fig. 5(b,d,e) yields of parent ions relative to total ions are plotted for bandwidth limited 100 fs pulses and pulses stretched to 350 fs or more. For 100 fs pulses, the butadiene parent ion production increases with decreasing intensity until 100% is attained below 2.5 × 1013 Wcm−2. A similar upward trend in parent ion yield with lower intensities is seen for butene, but for butane the yield is only about 10% and relatively independent of intensity. This suggests that at a wavelength of 800 nm it is not possible to ionise butane without delivering the small amount of excess energy needed to open up dissociation channels.
The longer pulse lengths make little difference to the parent yield for butadiene, but give lower yields in butene and butane. This demonstrates the importance of timescale in determining whether or not dissociation channels can be accessed. For molecules with large numbers of degrees of freedom, the electron hole in the radical cation can have many destinations for little additional energy. Therefore, a large number of low lying electronic excited states provide routes for resonantly coupling further energy into the molecule and hence opening up a number of dissociation pathways. As these states generally have different geometries compared to the neutral molecule, the resulting Franck–Condon overlaps are poor. However, since the cation is usually formed with additional vibrational energy, subsequent conformational changes open up allowed transitions to these levels. This explains why reduction in pulse length reduces the fraction of fragment ions seen for butene and butane. This general rule has also been noted in previous studies17 and consequently it can be expected that ultrashort pulses with durations less than the fastest vibrational periods (10–15 fs for a C–H stretch) will strongly promote formation of parent ions.
There have been some previous studies of these molecules in intense 800 nm laser fields. Palaniyappan et al.18 have generated mass spectra for n-butane with 45 fs pulses at intensities greater than 1014 Wcm−2. In agreement with our observations, these results show that intensity has little influence on the relative parent ion yield. Medhi Sharifi et al.4 have produced results for 1-butene with 175 fs pulses which show a less prominent parent peak compared with our results at an intensity of 6.5 × 1013 Wcm−2 (Fig. 3c). The shorter pulses in our experiment could explain this difference along with the fact that our detection efficiency does not decrease with mass unlike typical electron multiplier detectors.
1400nm
While 800 nm is the most convenient wavelength to use for femtosecond multi-photon ionisation, other wavelengths can be accessed through harmonic generation or optical parametric amplification. Resonantly enhanced two photon ionisation of active chromophores using the third harmonic (267 nm) has been demonstrated to be effective at generating the parent ions of peptides.19 For a wider range of species, it has been suggested that the mid-infrared wavelengths could reduce fragmentation compared to 800 nm pulses with similar parameters.20 If the photon energy is too high to excite any vibrational modes but sufficiently low to limit resonant excitation to electronic states of the cation, then less energy should be coupled into the molecule.To investigate the applicability of this hypothesis for the present molecules, in Fig. 6 we have compared 1-butene spectra obtained at 1400 nm with the 800 nm results at the same intensity. It can clearly be seen that the longer wavelength reduces the contribution of fragment ions, thus enhancing the parent ion yield. An even more dramatic increase is obtained for butane where the parent yield increases from 15% to 38%. In Table 2, these yields at an intensity of 2 × 1013 Wcm−2 are compared with that obtained from electron impact ionisation at 70 eV.
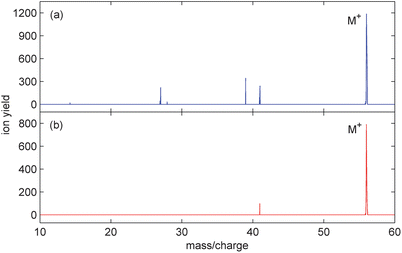 | ||
| Fig. 6 Mass spectra from 1-butene ionised with 100 fs pulses of intensity 2 × 1013 Wcm−2 and wavelength (a) 800 nm and (b) 1400 nm. | ||
Conclusions
From our results it is evident that an intense, femtosecond laser with appropriate parameters outperforms an electron beam as an ionisation method for a mass spectrometer. For studies of catalytic hydrogenation of 1,3-butadiene it would be particularly difficult to identify n-butane products by electron impact due to very low parent ion yields. While it is not possible to quote absolute ionisation efficiencies since the focussed pulse produces a wide range of intensities with different interaction volumes, our results show that tens of thousands of ions are produced per pulse for molecular targets with number densities of 1015 cm−3. This suggests that a single pulse is capable of generating sufficient ions for identification of chemicals with partial pressures less than 10−10 mbar.These characteristics along with the laser's pulsed operation, make it ideal for coupling to a time of flight mass spectrometer. With the advent of commercial high power, high repetition rate (10 kHz or greater) femtosecond lasers in the last few years, time-resolved mass spectra of catalytic reactions is now possible. At the Paul Scherrer Institute such analysis will be undertaken on a new apparatus which will use frequency modulated gas jets to investigate true kinetic timescales for catalysis. A high repetition rate femtosecond laser will enable experiments in low microsecond regimes, characteristic of catalytic product formation. This equates to three orders of magnitude improvement in time resolution, compared to the state-of-the-art equipment.21
For general application of femtosecond lasers in mass spectrometers used for chemical analysis, this work identifies pulse parameters which optimise yields of parent molecular ions. We have shown that there is a tendency to generate fewer fragment ions as the intensity is decreased but not necessarily for molecules where cation dissociation channels lie close to the ionisation threshold (e.g.butane).
We have also seen a strong increase in fragment ions as the laser pulse length is increased. We have attributed this to the molecule having a greater opportunity for absorbing further photons following nuclear relaxation into structures which provide greater Franck–Condon overlap with excited states. By using longer wavelengths we have also shown that access to these excited states is limited and results in further suppression of fragment peaks.
To summarise, femtosecond lasers are likely to find significant uses in mass spectrometry over the next few years. They provide high ionisation efficiency and depending on the pulse parameters, they can effectively break up molecules for structural analysis,22 or be optimised for parent molecular ion generation in high sensitivity chemical analysis. For the latter, we can recommend that the laser should ideally have the shortest possible pulse length, peak intensities below the saturation intensity for single ionisation, and wavelengths in the mid-infrared.
Acknowledgements
CRC acknowledges funding from an EPSRC Postdoctoral Fellowship, OK and RBK from the Leverhulme Trust, LB and MJD from the Department of Employment and Learning (NI). This work was carried out using the UFL2 system from the Laser Loan Pool, funded by EPSRC.References
- L. Hanley and R. Zimmerman, Anal. Chem., 2009, 81, 4174 CrossRef CAS.
- Y. Li and F. Qi, Acc. Chem. Res., 2010, 43, 68 CrossRef CAS.
- K. W. D. Ledingham and R. P. Singhal, Int. J. Mass Spectrom. Ion Processes, 1997, 163, 149 CrossRef CAS.
- S. Medhi Sharfi, A. Talebpour and S. L. Chin, Appl. Phys. B: Lasers Opt., 2008, 2008(91), 579 CrossRef.
- I. Pastirk, X. Zhu, V. V. Lozovoy and M. Dantus, Appl. Opt., 2007, 46, 4041 CrossRef.
- For example: D. Seth, A. Sarkar and F. T. T. Ng, Garry L. Rempel, Chem. Eng. Sci., 2007, 62, 4544 CAS.
- X. Zhang, F. X. L.i. Xamena and A. Corma, J. Catal., 2001, 265, 155 CrossRef.
- SIMION® ion optics software, SIS, Inc Search PubMed.
- J. B. Greenwood, et al. , Rev. Sci. Instrum., 2011, 82, 043103 CrossRef CAS.
- K. Yamakawa, et al. , Phys. Rev. Lett., 2004, 9(2), 123001 CrossRef.
- A. Becker and F. H. M. Faisal, J. Phys. B: At., Mol. Opt. Phys., 1999, 32, L335 CrossRef CAS.
- J. D. Alexander, et al. , Meas. Sci. Technol., 2010, 21, 045802 CrossRef.
- Values from: http://webbook.nist.gov/chemistry/.
- J. Muth-Böhm, et al. , Phys. Rev. Lett., 2000, 85, 2280 CrossRef.
- M. Lezius, et al. , J. Chem. Phys., 2002, 117, 1575 CrossRef CAS.
- L. V. Keldysh, Sov. Phys. JETP, 1965, 20, 1307 Search PubMed.
- V. V. Lozovoy, X. Zhu, T. C. Gunaratne, D. Ahmasi Harris, J. C. Shane and M. Dantus, J. Phys. Chem. A, 2008, 112, 3789 CrossRef CAS.
- S. Palaniyappan, et al. , Phys. Rev. A: At., Mol., Opt. Phys., 2010, 82, 043433 CrossRef.
- R. Weinkauf, P. Aicher, G. Wesley, J. Grotemeyer and E. W. Schlag, J. Phys. Chem., 1994, 98, 8381 CrossRef CAS.
- D. Willingham, A. Kucher and N. Winograd, Chem. Phys. Lett., 2009, 468, 264 CrossRef CAS.
- (a) R. Horn, G. Mestl, M. Thiede, F. C. Jentoft, P. M. Schmidt, B. Bewersdorf, R. Weber and R. Schlögl, Phys. Chem. Chem. Phys., 2004, 6, 4514 RSC; (b) G. McDougall Use of frequency response techniques to study the kinetics and identify active sites in metal catalysed oxidation reactions' 187th IRDG Meeting ( 2008) Glasgow University Search PubMed.
- C. L. Kalcic, T. C. Gunaratne, A. D. Jones, M. Dantus and G. E. Reid, J. Am. Chem. Soc., 2009, 131, 940 CrossRef CAS.
Footnote |
| † Paul Scherrer Inst, CH-5232 Villigen, Switzerland. |
| This journal is © The Royal Society of Chemistry 2012 |