DOI:
10.1039/C0SM00899K
(Paper)
Soft Matter, 2011,
7, 3092-3099
Influence of nano-viscosity and depletion interactions on cleavage of DNA by enzymes in glycerol and poly(ethylene glycol) solutions: qualitative analysis
Received
31st August 2010
, Accepted 8th November 2010
First published on 15th December 2010
Abstract
Biochemical reactions in living systems take place in an environment crowded by various macromolecules and ligands. Therefore experimental data obtained in buffer do not reflect in vivo conditions. We have used glycerol, poly(ethylene glycol) (PEG) 6000 and PEG 8 M solutions to investigate the influence of the crowded environment on cleavage of plasmid DNA by restriction enzyme HindIII. PEG 6000 solution can effectively slow down the cleavage process. However, neither PEG 8 M solution of the same viscosity as PEG 6000 solution nor glycerol solution of the same concentration as PEG 6000 solution slows the cleavage of DNA appreciably. The viscosity experienced by the biomolecules (here called nano-viscosity) and aggregation induced by the depletion interactions between DNA molecules in polymer solution (PEG 6000) are two factors responsible for slow cleavage of DNA. We have ruled out the change of pH and denaturation of HindIII as possible sources for the effect.
Introduction
Typical biochemical experiments done in buffers do not reflect in vivo conditions. In living cells, various macromolecules and lipids constitute up to 40% of the volume.1,2 Consequently, biological reactions in vivo inevitably occur in a crowded environment.3 Such environment strongly affects the stability of proteins,4,5 the diffusion coefficient of biomolecules,6 the activity of enzymes,7,8 and the association rate of proteins.9 Biochemistry in a crowded environment in vitro is a new emerging field of research which might give an insight into biochemical reactions in vivo.10 Many soft matter systems (e.g.polymer11 and micelle solutions6) can serve as paradigms of crowded environment mimicking the conditions inside of cells. In this manuscript we present a qualitative study of DNA cleavage by proteins in polymer (PEG) solutions and for comparison in low molecular mass agent (glycerol) solutions.
DNA cleavage by restriction enzyme HindIII is important in nucleic acid metabolism,12 protection against virus attack,13 and biotechnology.14 HindIII is a kind of type II restriction enzyme that digests the double-stranded DNA at the palindromic sequence AAGCTT. Although many studies have been done to investigate the structure, stability and function of type II restriction enzymes,15–17 only a few of them have been done in crowded environment.16,17 Previous studies showed that crowded environment had different influences on different enzymes. For example, Zimmerman and Pheiffer18 reported that crowded environment could increase the activity of DNA ligase. But Wenner and Bloomfield16 showed that crowded environment had negligible influence on the activity of EcoRV. These studies16,18 indicated a lack of universal rule describing enzymes activity in crowded environment. Although HindIII utilizes a similar mechanism of recognition and cleavage of DNA as other type II restriction enzymes,19 such as EcoRI,20 little information is available about cleavage of DNA by HindIII in crowded environment.
The viscosity is one of the characteristics of crowded environment that affects cleavage of DNA. From the Stokes–Sutherland–Einstein equation, an increase of viscosity decreases the diffusion coefficient of enzyme and DNA in the solution. As a result, according to the Smoluchowski theory this bioreaction is slowed down. However, the crowded environment, which is featured by large viscosity, does not always decrease diffusion coefficients of biomolecules appreciably. For example, some small proteins move fast in various cell compartments such as nucleus,21cytoplasm,22 and mitochondria,23,24 despite high viscosity in these systems. According to the Stokes–Sutherland–Einstein equation, this fast diffusion indicates that the proteins experience a viscosity much smaller than the macro-viscosity measured by a standard rheometer.6 We have provided an explanation for this phenomenon in our recent study of tracers (proteins, dyes) mobility in polymer solutions.25 We have connected the nano-viscosity experienced by the tracers in polymer solutions to the size of the polymer molecules. We have shown that when the diameter of the tracer, d, is bigger than the polymer radius of gyration Rg, the tracers experience the macro-viscosity of the polymer solution. On the other hand, when d is much smaller than Rg the tracers experience a nano-viscosity much smaller than the macro-viscosity.25 Therefore we made a clear distinction between macro- and nano-viscosity in our study of cleavage of DNA in polymer solution. To the best of our knowledge, this is the first research which makes such distinction between nano- and macro-viscosity in connection with the cleavage of DNA in polymer solutions.
Another characteristics of crowded environment which affect the cleavage of DNA are depletion interactions.26–28 PEG can induce DNA collapse into a compacted structure through attractive depletion interaction between DNA fragments.29,30 Depletion interactions are caused by the osmotic pressure which arises when two DNA molecules are in close proximity, closer than the radius of gyration of the polymer. In such a case, the space between DNA molecules is depleted from polymer coils and consequently an imbalance in osmotic pressure occurs. It leads to an effective attraction between DNA fragments of the same DNA molecule. The same interactions are responsible for the formation of large aggregates, when acting between different DNA molecules. For long DNA molecules monomolecular collapse dominates29 and for short DNA molecules aggregation of DNA molecules dominates.31 As a result DNA forms a compact state.29 The structural changes inside DNA can in principle strongly affect its interactions with enzymes and consequently affect the cleavage process of the DNA.
In this study, we investigate the influence of glycerol, PEG 6000 and PEG 8 M solutions on the cleavage of DNA by HindIII. We show by electrophoresis assay that PEG 6000 solution decreases the reaction rate more efficiently than glycerol solution of the same concentration or PEG 8 M solution of the same macro-viscosity. We measure the viscosity of PEG and glycerol solutions by dynamic light scattering (DLS) and a standard viscometer. We explain the DNA cleavage in PEG and glycerol solution by the concept of size dependent nano-viscosity. We also compare the size of DNA obtained by DLS measurement with that obtained by theoretical analysis and demonstrate formation of aggregates of plasmid DNA in PEG 6000 solution due to depletion interactions. We show that cleavage of DNA is extremely slow when aggregates appear. We rule out the change of pH and denaturation of HindIII as a source of slow cleavage of DNA.
Results and discussion
DNA cleavage in the crowded environment
We employed the electrophoresis assay32 to monitor the cleavage of DNA by HindIII in glycerol, PEG 6000 and PEG 8 M solutions respectively (Fig. 1). PUC19 plasmid DNA is composed of two components. Component I DNA is the supercoiled double-stranded DNA. Component II DNA is the loose circular DNA, which results from single-stranded cleavage of component I DNA. These components of DNA have different mobilities in the agarose gel and are presented as two distinguishable bands in electrophoresis assay. Restriction enzyme HindIII can cut double-stranded DNA at the palindromic sequence AAGCTT and transform PUC19 DNA into a linear form of DNA (component III). Component III DNA moves faster than component I and component II DNA in electrophoresis assay. The difference in mobility of all three components of the plasmid DNA is caused by the difference in their structure. We show in Fig. 1 that PEG 6000 solution can efficiently inhibit the cleavage of DNA, i.e. we find that a large amount of DNA is not changed to component III DNA in the presence of high concentration of PEG 6000 solution. On the contrary, PEG 8 M solution does not show any influence on DNA cleavage process. We also find that glycerol solution of the same concentration as the PEG 6000 solution also slows down the cleavage of DNA but with low efficiency. We note that PEG 6000 solution is characterized as crowded and viscous; glycerol solution is characterized as crowded but not viscous; and PEG 8 M solution is characterized as viscous but not crowded. In this study, we investigate the nanoscopic mechanism behind the unusual cleavage of DNA in the crowded environment.
 |
| | Fig. 1 The photograph of the gel electrophoresis assay performed after the cleavage of the DNA by HindIII in PEG 6000, glycerol and PEG 8 M solutions. The reaction time (1 h) was the same for all solutions. The photograph shows three separate bands for I, II, and III components of the plasmid DNA. Component III is the linear DNA which results from the cleavage process of circular DNA plasmid. This component has the highest mobility. Lane 1 is the control lane showing native DNA without any cleavage (same in all cases). Lanes 2 to 7 denote solutions with different PEG 6000 (a) or glycerol (b) concentrations i.e. 0, 7, 14, 21, 29 and 36 w/w% respectively. In the case of PEG 8 M (c) lanes 2 to 6 correspond to 0, 0.14, 0.49, 0.63 and 0.7 w/w% respectively. | |
Influence of viscosity on DNA cleavage process
The viscosity of crowded environment influences the association/dissociation rate of protein binding, equilibrium constant and reaction rate of biological reactions.10,11 We investigated the influence of viscosity on the cleavage of DNA. Glycerol, PEG 6000 and PEG 8 M solutions are three examples of crowded environments of different viscosity characteristics (Fig. 2). The viscosity of glycerol, PEG 6000 and PEG 8 M solutions increases with their concentration. However, the glycerol solution has much smaller viscosity than the PEG 6000 solution of the same concentration; while PEG 8 M solution has much smaller concentration than the PEG 6000 solution of the same viscosity. The viscosity of PEG solution grows25 with polymer concentration x as exp (3/2(x/x*)0.53). Here x* = Mp/(4/3πRg3NA) is the overlap concentration i.e. the concentration at which polymer's chains start to overlap. Mp is the molar mass of polymer, NA is Avogadro's number and Rg = 0.02Mp0.58 [nm] is the radius of gyration of PEG chain.33 The overlap concentration is 0.08 g cm−3 for PEG 6000 (average molecular mass Mp = 3461 Da) and 0.002 g cm−3 for PEG 8 M (average molecular mass Mp = 854096 Da). Consequently, the concentration of PEG 6000 solution is larger than that of PEG 8 M solution of the same viscosity.
 |
| | Fig. 2 Viscosity as a function of concentration of glycerol, PEG 6000 and PEG 8 M in TE solutions at 37 °C. | |
In glycerol and PEG 6000 solutions, the diffusion coefficient D of HindIII or DNA is inversely proportional to the viscosity η of the solution according to the Stokes–Sutherland–Einstein equation, D = kT/(6πηR) (see the discussion of the application of this equation at nanoscale25). Here k is the Boltzmann's constant and T is the temperature. The viscosity of polymer solutions is much larger than that of TE buffer, thus the diffusion coefficients of HindIII and DNA in crowded environment are much smaller than those in TE buffer. Small diffusion coefficient of HindIII and DNA increases the time for these reagents to encounter in the crowded environment. Consequently, the cleavage of DNA in crowded environment is slower than that in TE buffer (Fig. 1a and b).
The viscosity-determined, diffusion-limited theory well explains the slow cleavage of DNA in glycerol and PEG 6000 solution. But it cannot explain why PEG 8 M solution, which is also characterized by large viscosity, does not affect the cleavage of DNA (Fig. 1c). In order to explain this unusual phenomenon we employ the concept of nano-viscosity. We have shown previously25 that the diffusion of proteins in polymer solutions should be characterized by the size dependent nano-viscosity.25,34 We established experimentally that when the radius of gyration of the polymers, Rg, is larger than the diameter d of proteins, Rg > d, the proteins experience a nano-viscosity, given by:25
| | | ηnano = exp (b(d/ξ)a)η0 | (1) |
But when
Rg <
d, the
proteins experience a macro-viscosity, given by:
25| | | ηmacro = exp (b(Rg/ξ)a)η0 | (2) |
In
eqn (1) and
(2),
η0 is the viscosity of
water,
a and
b are constants of the order of 1 and
ξ is the concentration dependent correlation length (mesh size) of a
polymer network. Within a sphere of diameter
ξ all monomers belong to only one polymer chain. For PEG solutions
a = 0.7,
b = 1.5 and
ξ =
Rg(
x/
x*)
−0.75. For PEG 8 M the radius of gyration is 55.12 nm and for
x = 0.01 g cm
−3 (1 w/w% PEG 8 M solution) we find
ξ = 16.5 nm. The size of HindIII (estimated on the basis of its molecular mass 69.9 kDa) is
d = 5.6 nm. Now from
eqn (1) we find that the nano-viscosity experienced by HindIII in PEG 8 M solution is two times larger than
water viscosity
ηnano = 2
η0. Such small nano-viscosity means that HindIII moves relatively freely in PEG 8 M solution. That is why PEG 8 M does not affect the cleavage of DNA. Please note that the effective length of the DNA based on the equivalent rod model is 266 nm (estimated from the analysis presented in the Experimental section), which is much larger than
Rg = 55.12 nm of PEG 8 M and thus DNA can be treated as immobile in comparison to the motion of the enzyme of size
d = 5.6 nm.
The radius of gyration, Rg, for PEG 6000 is 2.26 nm and for glycerol is even smaller. That is why HindIII of size d = 5.6 nm > Rg experiences the macro-viscosity in these solutions. The time needed to cut DNA in these solutions should be proportional to the macro-viscosity and that is why the cleavage time grows with the concentration of glycerol or PEG 6000 according to eqn (2) (see Fig. 1 and 2).
Previous study16 concerning EcoRV kinetics in Ficoll solutions also mentioned that the slow diffusion of proteins caused by viscosity influences the cleavage of DNA. However, the authors did not relate the cleavage of DNA to the nano-viscosity. We showed in our study that it is the nano-viscosity rather than the macro-viscosity that affects the cleavage of DNA in the crowded environment.
Influence of depletion interactions on DNA cleavage
We observe that the cleavage process at high concentration PEG 6000 is too slow to be explained by the changes of the nano-viscosity or macro-viscosity. The viscosity of 14 w/w% PEG 6000 is about 10 times larger than that of TE buffer. According to the Stokes–Sutherland–Einstein equation, the diffusion coefficient of HindIII in 14 w/w% PEG 6000 is about 10 times smaller than that in TE buffer. Because the cleavage of DNA in TE buffer solution is completed in 10 min (Fig. 3), HindIII should complete the cleavage in the 14 w/w% PEG 6000 solution in 100 min. However, the cleavage process is not completed in 1080 min (Fig. 3), showing that there were other factors influencing the cleavage of DNA in PEG 6000 solution.
 |
| | Fig. 3 The photograph of the gel electrophoresis assay presenting an influence of the incubation (reaction) time on DNA cleavage process in PEG 6000 solutions. Lane 1 is native DNA. The concentration of PEG 6000 solution from lane 2 to lane 7 is 0, 7, 14, 21, 29 and 36 wt% respectively. The incubation time at 37 °C is 10 min (a) and 18 h (b) respectively. The I, II and III bands in lane 4 in (a) and (b) indicate that the process is not completed in 18 h. Intensity for the third component (III) is similar in (a) and (b) showing that the process is extremely slow. | |
The cleavage of DNA in crowded environment involves various nonspecific interactions of biomolecules with solution constituents apart from the viscosity. We have already mentioned in the Introduction the depletion interaction between DNA molecules in polymer solutions.29,30 The influence of this interaction on reaction rates was strong in our solutions, although indirect. The origin of depletion interactions can be traced back to the conformational entropy of polymer chains.27,35 The center of mass of a polymer molecule cannot get closer to the DNA fragment than the certain characteristic distance because it would result in a decrease of its conformational entropy. This distance is proportional to the sum of the radius of gyration of a polymer, Rg, and a radius of a DNA fragment, r. If two DNA fragments are close to each other, separated by a distance smaller than 2(r + Rg), a polymer cannot enter between them. This depletion zone (zone depleted from polymers) causes an imbalance of osmotic pressures (induced by polymers outside the zone between the DNA fragments). The osmotic pressure pushes the DNA fragments together and collapses DNA into a compacted structure. If the DNA fragments belong to the same DNA molecule we call it condensation.29,30 If the DNA fragments belong to different DNA molecules we call it aggregation.31DNA condensation decreases the volume of DNA molecules and increases their diffusion coefficient; while DNA aggregation increases the volume of DNA molecules and decreases their diffusion coefficient. Both processes compact the structure of DNA and spatially inhibit the approach of HindIII to its target DNA. As a result the cleavage of DNA is slowed down.
In order to prove aggregation of DNA molecules in PEG 6000 solution at high concentration we have performed DLS experiments and theoretical analysis for the diffusion coefficients of the plasmid DNA (Experimental section for details). The theoretical analysis, according to Seils and Pecora,36 uses an equivalent rod as a model for the plasmid DNA to compute its diffusion coefficient. This model is fitted to the experimental data in TE buffer (Fig. 4) showing perfect agreement between theory and experiment. However, in polymer solution theoretical model and experimental results disagree by a factor of 2 to 4. We observe that DNA molecules diffuse slower than expected at both 25 °C and 37 °C (Fig. 4). For example the DNA molecules in 36 w/w% PEG 6000 solution at 37 °C diffuse 2.3 times slower than expected on the basis of the model and the value of the macro-viscosity. The only way to reconcile these data with the theoretical model is the large increase (2.3 times) of the size of DNA. Such increase in the linear size indicates aggregation of many DNA coils. Because the volume of the aggregate scales as the cube of the size we can give a lower limit of the number of DNA coils in the aggregate i.e.N = 2.33 = 12. Such aggregation is possible because DNA coils are not separated by large distances in the solution. The concentration of DNA is about 23.2 nmol L−1. So the average distance between DNA molecules is about 0.4 µm. The contour length of pEGFP-C1 DNA is about 1.6 µm and the diameter of DNA is about 2 nm. The average distance between DNA molecules and the size of DNA molecules are comparable and thus aggregation is probable. Both intermolecular DNA aggregation and intra-molecular DNA condensation can be induced by the depletion interactions. We think that DNA aggregation is additionally accompanied by DNA condensation, but we do not have any experimental verification of this phenomenon.
 |
| | Fig. 4 Diffusion coefficient of plasmid DNA measured by DLSversus the theoretical data at 25 °C (a) and 37 °C (b). Disagreement between theory (calculated for a single molecule of native DNA) and experiment indicates aggregation of plasmid DNA in polymer (PEG 6000) solution. | |
Actually aggregation occurs also at very small concentrations of DNA (2.5 nM) for short DNA fragments (66 bp) as we checked using the fluorescence correlation spectroscopy (FCS) (Fig. 5). The diffusion time of DNA fragments in the confocal volume in TE buffer is about 0.2 ms. But we cannot establish a proper FCS curve in 36% PEG 6000 solution because we only observe large objects in the confocal volume. From Fig. 5, we find that the diffusion time of the particles through the confocal volume is close to few seconds. But the viscosity of 36% PEG 6000 solution is only 50 times larger than that of the buffer. Taking the viscosity into consideration, the size of particles estimated from the diffusion time is about 100 times larger than that of a single DNA fragment, indicating appearance of large aggregates.
 |
| | Fig. 5 Intensity of the fluorescence for labeled 66 bp DNA fragments in 36 w/w% PEG 6000 solution at 37 °C showing formation of large aggregates of DNA molecules. Strongly elevated fluorescence (vertical axis) lasts for few seconds. Such long time is due to very large objects incompatible with short DNA fragments. Short fragments should move across confocal volume in a millisecond time scale. Long time (seconds) of diffusion across the confocal volume can only be explained by the aggregation of DNA fragments. | |
Influence of pH on HindIII activity
It is often observed that pH has tremendous influence on biochemical reactions. Therefore we investigate whether the slow cleavage of DNA is caused by the change of pH in glycerol and PEG solutions (Fig. 6). The buffer has a pH = 7.5; for 50% PEG 6000 pH = 6 and for 50% glycerol pH = 7.2. PEG 8 M does not change the pH value obviously when its concentration is no more than 1%. We have performed DNA cleavage in buffers of different pH values from 7.5 to 4 and observed no visible changes in the process (Fig. 6). Consequently the cleavage of DNA in our study should not be influenced by the changes of pH in the crowded environment.
 |
| | Fig. 6 The pH values of PEG 6000 solution, glycerol solution and PEG 8 M solution as a function of the concentration. The inset image shows the photograph of the gel electrophoresis assay done in buffers of different pHs. Lane 1 is native DNA at pH 7.5 as a control. The pH values of the buffer solution for lane 2 to lane 7 are 7.5, 7.2, 6.7, 6, 5 and 4 respectively. | |
Influence of glycerol and PEG 6000 on HindIII activity
Large molecules, such as sodium dodecyl sulfate (SDS), can dramatically decrease the activity of enzymes by changing the secondary structure of proteins.37 We showed that neither glycerol nor PEG 6000 deactivates HindIII (Fig. 7). We incubated the HindIII in concentrated glycerol solution or PEG 6000 solution for 3 h. Then we used this mixture to cleave DNA. Electrophoresis assay showed that HindIII cleaved DNA after incubation for 3 h. This experiment proves that activity of HindIII is not influenced by PEG or glycerol.
 |
| | Fig. 7 (a) DNA cleavage by HindIII after HindIII incubation in glycerol solutions. Lane 1 is native DNA. Lane 2 is native DNA cleaved by HindIII. Lane 3 shows DNA cleavage by HindIII, after HindIII incubation in 30% glycerol solution for 3 h. Lane 4 shows DNA cleavage by HindIII, after HindIII incubation in 36% glycerol solution for 3 h. (b) DNA cleavage by HindIII after HindIII incubation in PEG 6000 solutions. Lane 1 is native DNA. Lane 2 is native DNA cleaved by HindIII. Lane 3 is DNA cleavage by HindIII, after HindIII incubation in 30% PEG 6000 solution for 3 h. Lane 4 is DNA cleavage by HindIII, after HindIII incubation in 36% PEG 6000 solution for 3 h. | |
We also find that when the reaction time is prolonged, more and more DNA is cleaved in the presence of glycerol solution (Fig. 8). This experiment indicates that HindIII is active in glycerol solution. Many researches11,38 also indicate that these crowding agents do not deactivate HindIII.
 |
| | Fig. 8
DNA cleavage in glycerol solution for (a) 10, (b) 20, (c) 30, (d) 40, (e) 50 and (f) 60 min. The concentrations of glycerol from lane 1 to lane 6 are 0, 7, 14, 21, 29 and 36 wt% respectively. It is clear that the glycerol slows down the cleavage, but does not stop it altogether. | |
Conclusion
We investigate the DNA cleavage process by HindIII in crowded environments composed of glycerol, PEG 6000 and PEG 8 M. We find that PEG 6000 solution could effectively slow down the cleavage process. However, neither PEG 8 M solution of the same viscosity as PEG 6000 solution nor glycerol solution of the same concentration as PEG 6000 solution slows down the cleavage of DNA appreciably. We show that the viscosity experienced by the biomolecules (nano-viscosity) is responsible for this effect. Large nano-viscosity of crowded environment (as in PEG 6000) decreases the diffusion coefficient of DNA and HindIII and thus reduces the frequency of encounters between DNA and HindIII. As a consequence the cleavage of DNA slows down. On the contrary PEG 8 M of high macro-viscosity does not influence the cleavage process because the nano-viscosity for HindIII is close to that of water. Additionally the depletion interactions between DNA also slow cleavage of DNA by inducing aggregation of DNA coils. When the concentration of PEG 6000 is high, DNA molecules form big aggregates due to depletion interactions, inhibiting the approach of HindIII to its target DNA. We rule out the change of pH and denaturation of HindIII as possible sources for slowing down the process. Our research indicates a convenient way to control the DNA cleavage rate by performing this reaction in a suitably chosen crowded environment.
Experimental section
General
Glycerol was purchased from Chempur. PEG 6000 and PEG 8 M were purchased from Fluka. The distribution of molecular weight of PEG was measured by the gel permeation chromatography. The number average molecular weight (Mn) of PEG 6000 was 3461 Da and of PEG 8 M was 854![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 096 Da (Fig. 9). Plasmid DNA PUC19 (2686 base pairs) was purchased from Bioron and stored in TE buffer (10 mmol L−1 Tris–HCl, 1 mmol L−1EDTA, pH 7.5). Plasmid DNA pEGFP-C1 was purified from Escherichia coli by QIAGEN plasmid maxi kit (QIAGEN, Germany) and was stored in TE buffer. The concentration and purity of pEGFP-C1 were measured by using a UV-spectrometer (MultiSpec-1501, Shimadzu). HindIII was purchased from Fermentas. The electrophoresis assay system was purchased from Bio-rad.
096 Da (Fig. 9). Plasmid DNA PUC19 (2686 base pairs) was purchased from Bioron and stored in TE buffer (10 mmol L−1 Tris–HCl, 1 mmol L−1EDTA, pH 7.5). Plasmid DNA pEGFP-C1 was purified from Escherichia coli by QIAGEN plasmid maxi kit (QIAGEN, Germany) and was stored in TE buffer. The concentration and purity of pEGFP-C1 were measured by using a UV-spectrometer (MultiSpec-1501, Shimadzu). HindIII was purchased from Fermentas. The electrophoresis assay system was purchased from Bio-rad.
 |
| | Fig. 9 Distribution of molecular weights for PEG 6000 and PEG 8 M obtained using gel permeation chromatography (GPC). For PEG 6000 the average mass is Mn = 3461 Da and for PEG 8 M Mn = 854096 Da. These values were used to calculate the radius of gyration for PEG coils. | |
Electrophoresis assay for DNA cleavage by HindIII in crowded environments
In order to maintain the activity of DNA and HindIII, TE buffer was used for all crowding agents. The concentrations of glycerol and PEG 6000 solutions were prepared as 10, 20, 30, 40 and 50 w/w% respectively. The concentrations of PEG 8 M solutions were 0.2, 0.5, 0.7, 0.9 and 1 w/w% respectively. We prepared the loading samples by mixing 2 µL of plasmid DNA PUC19 solution, 14 µL of PEG 6000 solution, 2 µL of HindIII solution (10 u µL−1) and 2 µL of HindIII buffer solution (10× Buffer R: 100 mmol L−1 Tris–HCl, 100 mmol L−1MgCl2, 1 mol L−1KCl, 1 mg mL−1 BSA, pH 8.5). The concentration of DNA solution was 200 ng µL−1. Consequently, the concentrations of PEG 6000 in the reaction mixture were 0, 7, 14, 21, 29 and 36 w/w% respectively. The native DNA was used as a control. The samples were incubated at 37 °C for 1 h and then investigated by the electrophoresis assay. The agarose gel (0.8%) containing 1 µg mL−1ethidium bromide was used in the electrophoresis assays. The gel was observed on a UV transilluminator (Biogenet) and the images were recorded by a digital camera. Cleavage of DNA in the glycerol and PEG 8 M solutions was performed in the same way as in PEG 6000 solution. The concentrations of glycerol in the reaction mixture were 0, 7, 14, 21, 29 and 36 w/w% respectively. The concentrations of PEG 8 M in the reaction mixture were 0, 0.14, 0.49, 0.63 and 0.7 w/w% respectively.
Viscosity measurements
The viscosities of glycerol and PEG 6000 solutions were measured by DLS using a BI-200SM Goniometer (Brookhaven Instruments Corp.) equipped with Argon Ion Stabilite 2017 laser. Nanobeads were purchased from Polysciences Inc. as tracers for viscosity measurement. The diameter of the beads was 88.08 nm on average. The solutions were filtered before measurement. The refractive index of the liquid solution for the DLS measurements was measured on a standard Abbe refractometer (Carl-Zeiss, Germany). We measured the viscosity of PEG 8 M by a standard viscometer KF10 (Rheotec Germany). The temperature of all samples was set at 37 °C.
Measurement of the diffusion coefficient of plasmid DNA in PEG 6000 solution by DLS
We used plasmid DNA PEGFP-C1 (4.7k base pairs) in DLS measurement. Both 7 and 36 w/w% PEG 6000 solutions (in TE buffer) were used. The concentration of plasmid DNA was 72 ng µL−1. We also added the same concentration of enzyme buffer into the solution to mimic the cleavage system. TE buffer containing the same amount of plasmid DNA was used as a control. DLS measurement was carried out using a BI-200SM Goniometer at both 25 °C and 37 °C.
The experimental results were fitted according to the typical DLS procedure. The characteristic diffusion time for DNA, τ0, and PEG 6000, τ0p, is obtained by evaluating the time-dependent homodyne autocorrelation function.
| | | g(q,τ) = 〈I(q,t)I(q,t + τ)〉 | (3) |
where
I(
q,
t) is the intensity of light scattered from the samples at time
t and at the wavevector
q. Here, the modulus of the scattering vector of the optical arrangement,
q, is given by
| |  | (4) |
where
n is the refractive index of the liquid solution measured by a standard Abbe refractometer (Carl-Zeiss, Germany),
λ is the wavelength of the laser in vacuum and
θ is the scattering angle. The autocorrelation function
g(
q,
t) from
eqn (3) for one diffusion mode is represented by a single exponential type of decay function for DNA in TE buffer solution,
| | | g(q,τ) = A + B [exp (−τ/τ0)]2, | (5) |
or a double exponential type of decay function for DNA in PEG 6000 solution,
| | | g(q,τ) = A + [B exp (−τ/τ0) + C exp (−τ/τ0P)]2 | (6) |
where
A,
B and
C are experimental constants. The inverse of the characteristic time in
eqn (5) and
(6) depends on
q as follows:
| |  | (7) |
Here
D is the diffusion coefficient, which does not depend on
q. For each sample we measured the autocorrelation function using five different scattering angles (
q wavevectors) and used
eqn (7) to determine
D.
Calculation of the diffusion coefficient of DNA using an equivalent rod model
According to the method developed by Seils and Pecora,36 we approximated shape of the supercoiled plasmid DNA by an equivalent rigid cylinder. The dimensions of the cylinder are calculated on the basis of the DNA superhelix in the following way. The length of this cylinder is half of the contour length of DNA, l, corrected for the helical pitch:| | 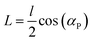 | (8) |
In eqn (8), αp is the pitch angle of the superhelix and L is the length of the cylinder. The diameter of the cylinder d is defined aswhere d0 represents the diameter of the relaxed DNA and r is the radius of helix.| | 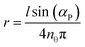 | (10) |
In eqn (10), nθ is the number of helical turns.where nb is the number of base pairs in DNA, nh is the number of base pairs for one turn, and δb is the number of superhelical turns per base pair.
For pEGFP-C1, we substituted following Seils and Pecora36αP = 55°, nb = 4700, nh = 10, δb = 0.079, and l = 1626.2 nm and we got the equivalent cylinder dimension: L = 466 nm and d = 13.4 nm. For PUC19, αP = 55°, nb = 2686, nh = 10, δb = 0.079, and l = 929.4 nm and we got the equivalent cylinder dimension: L = 266 nm and d = 13.4 nm.
For the equivalent cylinder, the translational self-diffusion coefficient D0 can be expressed as
| |  | (12) |
where
D⊥ describes the diffusion perpendicular to the long axis of the cylinder and
D‖ describes the diffusion parallel to the long axis of the cylinder.
| |  | (13) |
| |  | (14) |
In
eqn (13) and
(14),
kB is the Boltzmann constant,
T is the absolute temperature, and
ν⊥ and
ν‖ are “end-effect corrections”.
| | | v⊥ = 0.839 + 0.185/ρ + 0.233/ρ2 | (15) |
| | | v‖ = −0.207 + 0.980/ρ − 0.133/ρ2 | (16) |
In
eqn (15) and
(16)ρ is the axial ratio.
Electrophoresis assay for DNA cleavage by HindIII at different pHs
We adjusted the pH of TE buffer solutions to 7.5, 7.2, 6.7, 6, 5 and 4 respectively. The pH values of the solutions were measured at room temperature. The concentration of DNA solution was 200 ng µL−1. We prepared the samples by mixing together 14 µL of these TE buffer solutions, 2 µL of DNA solution, 2 µL of HindIII solution (10 u µL−1) and 2 µL of HindIII buffer solution. The mixed samples were incubated at 37 °C for 1 h and then investigated by electrophoresis assay. The native DNA was used as a control.
Electrophoresis assay for the activity of HindIII in glycerol and PEG 6000 solutions
We mixed together 4 µL of HindIII solution (10 u µL−1), 2 µL of HindIII buffer solution and 14 µL of glycerol or PEG 6000 solution. The concentration of glycerol in the final mixture was 30 and 36 w/w% respectively. The concentration of PEG 6000 in the final mixture was 30 and 36 w/w% respectively. The mixture was incubated at 37 °C for 3 h. Then 2 µL of the mixture was mixed with 2 µL of HindIII buffer solution, 2 µL of DNA solution and 14 µL of TE buffer solution, followed by incubation at 37 °C for 1 h. The samples were investigated by the electrophoresis assay in the same way as mentioned above.
FCS setup for the study of aggregation of short DNA fragments
FCS measurement was used to monitor the diffusion of DNA fragment in PEG 6000 solutions. We used a commercially available NIKON EZ-C1 confocal microscope equipped with Pico Harp 800 FCS setup made by PicoQuant for FCS measurement. We used a water immersion objective with numerical aperture equal to 1.2 and 60× magnification. Laser power was set on constant level of 35 µW. We used a HeNe laser of wavelength 543 nm. The ATTO550 labeled 66 bp DNA fragment was purchased from Eurofins (Fig. 10). We mixed together 1 µL of DNA fragment solution (500 nM), 20 µL of HindIII buffer solution and 180 µL of PEG 6000 solution. The concentration of PEG 6000 in the final mixture was 36 w/w%. The concentration of DNA fragment in the final mixture was 2.5 nM. DNA fragment in TE buffer was used as a control. We used an Okolab incubator to keep the temperature constantly at 37 °C. The FCS curve of control was fitted using a traditional method25 to get the diffusion time in the confocal volume.
 |
| | Fig. 10 Sequence of the double-stranded DNA fragment with ATTO550 labels. | |
Acknowledgements
The work was supported by the Project operated within the Foundation for Polish Science Team Programme co-financed by the EU “European Regional Development Fund” TEAM/2008-2/2 and also by Research Grant from the Human Frontier Science Program Organization, N.Z. We acknowledge a PhD scholarship from the President of the Polish Academy of Sciences and RH Mistrz grant from FNP.
References
- M. T. Record, E. S. Courtenay, D. S. Cayley and H. J. Guttman, Trends Biochem. Sci., 1998, 23, 143–148 CrossRef CAS.
- G. Rivas, F. Ferrone and J. Herzfeld, EMBO Rep., 2004, 5, 23–27 CrossRef CAS.
- C. P. Brangwynne, G. H. Koenderink, F. C. MacKintosh and D. A. Weitz, J. Cell Biol., 2008, 183, 583–587 CrossRef CAS.
- L. Stagg, S. Q. Zhang, M. S. Cheung and P. Wittung-Stafshede, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18976–18981 CrossRef CAS.
- K. Sasahara, P. McPhie and A. P. Minton, J. Mol. Biol., 2003, 326, 1227–1237 CrossRef CAS.
- J. Szymanski, A. Patkowski, A. Wilk, P. Garstecki and R. Holyst, J. Phys. Chem. B, 2006, 110, 25593–25597 CrossRef.
- B. R. Somalinga and R. P. Roy, J. Biol. Chem., 2002, 277, 43253–43261 CrossRef CAS.
- B. K. Derham and J. J. Harding, Biochim. Biophys. Acta, Proteins Proteomics, 2006, 1764, 1000–1006 CrossRef CAS.
- A. P. Minton and J. Wilf, Biochemistry, 1981, 20, 4821–4826 CrossRef CAS.
- Y. Phillip, E. Sherman, G. Haran and G. Schreiber, Biophys. J., 2009, 97, 875–885 CrossRef CAS.
- Y. Y. Kuttner, N. Kozer, E. Segal, G. Schreiber and G. Haran, J. Am. Chem. Soc., 2005, 127, 15138–15144 CrossRef CAS.
- M. Napirei, H. Karsunky, B. Zevnik, H. Stephan, H. G. Mannherz and T. Moroy, Nat. Genet., 2000, 25, 177–181 CrossRef CAS.
- N. Watanabe, Y. Takasaki, C. Sato, S. Ando and I. Tanaka, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 1326–1333 CrossRef.
- L. P. Cramer, L. J. Briggs and H. R. Dawe, Cell Motil. Cytoskeleton, 2002, 51, 27–38 CrossRef CAS.
- E. H. Serpersu, D. Shortle and A. S. Mildvan, Biochemistry, 1987, 26, 1289–1300 CrossRef CAS.
- J. R. Wenner and V. A. Bloomfield, Biophys. J., 1999, 77, 3234–3241 CrossRef CAS.
- Y. Sasaki, D. Miyoshi and N. Sugimoto, Nucleic Acids Res., 2007, 35, 4086–4093 CrossRef CAS.
- S. B. Zimmerman and B. H. Pheiffer, Proc. Natl. Acad. Sci. U. S. A., 1983, 80, 5852–5856 CrossRef CAS.
- D. H. Tang, S. Ando, Y. Takasaki and J. Tadano, Protein Eng., 2000, 13, 283–289 CrossRef CAS.
- U. Kettling, A. Koltermann, P. Schwille and M. Eigen, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 1416–1420 CrossRef CAS.
- R. D. Phair and T. Misteli, Nature, 2000, 404, 604–609 CrossRef CAS.
- M. B. Elowitz, M. G. Surette, P. E. Wolf, J. B. Stock and S. Leibler, J. Bacteriol., 1999, 181, 197–203 CAS.
- A. Partikian, B. Olveczky, R. Swaminathan, Y. X. Li and A. S. Verkman, J. Cell Biol., 1998, 140, 821–829 CrossRef CAS.
- A. Partikian, B. P. Olveczky, R. Swaminathan and A. S. Verkman, Biophys. J., 1998, 74, A382–A382.
- R. Holyst, A. Bielejewska, J. Szymanski, A. Wilk, A. Patkowski, J. Gapinski, A. Zywocinski, T. Kalwarczyk, E. Kalwarczyk, M. Tabaka, N. Ziebacz and S. A. Wieczorek, Phys. Chem. Chem. Phys., 2009, 11, 9025–9032 RSC.
- R. Tuinier and H. N. W. Lekkerkerker, Eur. Phys. J. E: Soft Matter Biol. Phys., 2001, 6, 129–132 Search PubMed.
- A. Vrij, Pure Appl. Chem., 1976, 48, 471–483 CrossRef CAS.
- S. Asakura and F. Oosawa, J. Chem. Phys., 1954, 22, 1255–1256 CAS.
- V. V. Vasilevskaya, A. R. Khokhlov, Y. Matsuzawa and K. Yoshikawa, J. Chem. Phys., 1995, 102, 6595–6602 CrossRef CAS.
- M. Kojima, K. Kubo and K. Yoshikawa, J. Chem. Phys., 2006, 124, 024902 CrossRef CAS.
- J. E. Ramos, Jr, R. de Vries and J. Ruggiero Neto, J. Phys. Chem. B, 2005, 109, 23661–23665 CrossRef.
- X. D. Chen, Y. Zhou, P. Qu and X. S. Zhao, J. Am. Chem. Soc., 2008, 130, 16947–16952 CrossRef CAS.
- K. Devanand and J. C. Selser, Macromolecules, 1991, 24, 5943–5947 CrossRef CAS.
- J. Liu, D. P. Cao and L. Q. Zhang, J. Phys. Chem. C, 2008, 112, 6653–6661 CrossRef CAS.
- F. Oosawa and S. Asakura, J. Chem. Phys., 1954, 22, 1255–1255 CAS.
- J. Seils and R. Pecora, Macromolecules, 1995, 28, 661–673 CrossRef CAS.
- C. Michaux, J. Pouyez, J. Wouters and G. G. Prive, BMC Struct. Biol., 2008, 8, 29 CrossRef.
- R. Grandori, I. Matecko, P. Mayr and N. Muller, J. Mass Spectrom., 2001, 36, 918–922 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.