Tracking gold acetylides in gold(I)-catalyzed cycloisomerization reactions of enynes†
Antoine
Simonneau
a,
Florian
Jaroschik
a,
Denis
Lesage
a,
Magdalena
Karanik
a,
Régis
Guillot
b,
Max
Malacria
a,
Jean-Claude
Tabet
a,
Jean-Philippe
Goddard
a,
Louis
Fensterbank
*a,
Vincent
Gandon
*b and
Yves
Gimbert
*c
aInstitut Parisien de Chimie Moléculaire (UMR CNRS 7201) - FR 2769 UPMC univ Paris 06, C. 229, 4 Place Jussieu, 75005, Paris, France. E-mail: louis.fensterbank@upmc.fr; Fax: +33 (0)1 44 27 73 60; Tel: +33 (0)1 44 27 70 68
bICMMO (UMR CNRS 8182), Université Paris-Sud 11, 91405, Orsay cedex, France. E-mail: vincent.gandon@u-psud.fr; Fax: +33 (0)1 69 15 47 47; Tel: +33 (0)1 69 15 39 31
cDCM (UMR CNRS 5250), ICMG-FR 2607, BP 53, 38041, Grenoble cedex 9, France. E-mail: yves.gimbert@ujf-grenoble.fr; Fax: +33 (0)4 76 51 44 94; Tel: +33 (0)4 76 51 44 26
First published on 9th September 2011
Abstract
The intermediacy of gold acetylides in the gold(I)-catalyzed cycloisomerization of enynes was questioned. While dinuclear gold complexes are observed under electrospray ionization conditions, the solution reactivity of gold acetylides also leads to the conclusion of their high affinity for the second coordination of a gold moiety, leading to dinuclear gold complexes. However, the involvement of gold acetylides and the corresponding diaurated species in the elementary steps of cycloisomerization mechanisms of enynes appears unlikely.
Gold-catalyzed reactions have lately emerged as a very powerful tool in organic synthesis.1 Although they have been increasingly documented, the true nature of the intermediates involved in these reactions remains quite uncertain.2 Mechanistic scenarios have been postulated, yet characterizations of putative intermediates that could support these hypotheses are still scarce3 and often lead to surprises. For instance, the formation of gem-diaurated species during gold(I)-catalyzed nucleophilic addition to alkynes and allenes has been recently experimentally demonstrated (Scheme 1, eqns (1) and (2)).4 Compelling evidence that gold-complexes of terminal alkynes can evolve into polynuclear species has also been provided.5 On the basis of deuterium-labeling experiments and DFT computations, it was also suggested that gold-catalyzed transformations involving terminal alkynes could involve, as elementary mechanistic steps, the formation of gold acetylides and the corresponding gem-diaurated species (eqn (3)).6
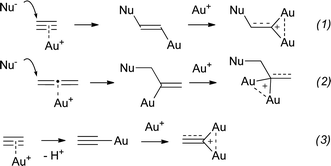 | ||
| Scheme 1 The prototypical formation of gem-diaurated species. | ||
Although in the case of the allenes depicted in eqn (2), the formation of the dinuclear complex was attributed to a catalyst resting state, diaurated species have also been considered as reactive intermediates.7 This was the case for the (Ph3PAu)3O+-catalyzed cycloisomerization of 1,5-allenynes, for which computations supported a dual activation mechanism, as shown in Scheme 2.6 Rather than a typical Au-triggered nucleophilic attack of the allene onto the complexed triple bond (pathway (i)), the 5-exo-dig cyclization is preceded by the formation of a gold acetylide and the corresponding diaurated species (pathway (ii)).
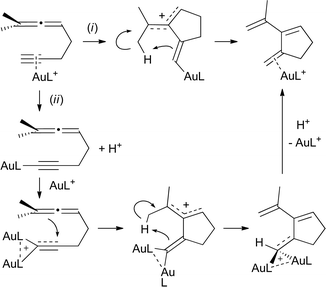 | ||
| Scheme 2 Monaurated (i) vs. diaurated (ii) mechanistic proposals for the gold(I)-catalyzed cycloisomerization of 1,5-enynes involving diaurated species. | ||
This type of reaction can be categorized within the vast area of 1,n-enyne cycloisomerizations, which has attracted considerable attention due to the high versatility of the process and its exceptional synthetic potential.8,9 We decided to check the validity of the dual activation mechanism depicted in Scheme 2 for the cycloisomerization of widely used 1,6-enynes. Using a combined approach involving mass spectrometry/solution reactivity/theoretical treatment,10 we herein provide new mechanistic elements.
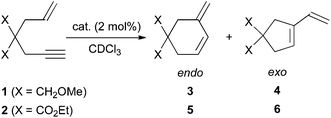
As a background for our study, we investigated the reactivity of enynes 1 and 2 with three cationic gold(I)-catalysts A,11B12 and C,13 which resulted in the very major formation of the 6-membered ring derivatives 3 and 5, accompanied by the formal metathesis products 4 and 6,14 respectively (Table 1).
|
|
||||
|---|---|---|---|---|
| a 60% of starting material recovered, no reaction at RT. b 55% of starting material recovered. c 38% of starting material recovered, no reaction at RT. | ||||
| Entry | Substrate | Cat. | Conditions | % Yield(endo:exo) |
| 1 | 1 | A | RT, 1 h | 87(20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
| 2 | 1 | B | reflux, 7 h | 35(1:0)a |
| 3 | 1 | C | RT, 1 h | quant. (5:1) |
| 4 | 2 | A | RT, 3 d | 38(6:1)b |
| 5 | 2 | B | reflux, 15 h | 47(4:1)c |
| 6 | 2 | C | RT,1 h | 95(1.6:1) |
|
||||

Then, we engaged enyne 1 with A–C in mass spectrometry conditions (Scheme 3, see also Figs S1–3 in the supporting information†). A solution of 1 and catalysts A–C in CDCl3 was subject to electrospray ionization (ESI) in positive mode and the resulting adduct ions were analyzed in a modified triple-quadrupole instrument.15 After mixing 1 with A or B, a peak corresponding to the formula [1–H + (Ph3PAu)2]+ (1-Au2) was found at m/z 1099.16 With 1 and C, the same type of adduct of formula [1–H + ((biphenyl-2-yl)(t-Bu)2PAu)2]+ (1′-Au22) was recorded at m/z 1171. The observation of these ions suggests the formation of gold acetylides, on which a second gold entity coordinates, thus creating gold dinuclear species. This was further corroborated by submitting the independently synthesized gold acetylide 1-Au17,18 to A–C or AgSbF6, leading to the expected peaks of the corresponding dinuclear species.19 The latter were next fragmented by Collision Induced Dissociation (CID), giving rise to cations bearing all of the phosphines. Therefore, in all cases, the same neutral counterpart of formula [1–H + Au] (loss of 378) was formed during CID. Because structural pieces of information could not be gleaned from fragment analysis, we carried out DFT computations at the B3LYP/LANL2DZ(Au)/6-31G(d,p) level of theory using hept-1-en-6-yne and PH3 as models (Scheme 4). From the dissociation free energies, one can notice that the naked gold acetylide or the related chelate require prohibitive energies to form under CID conditions. Thus, the neutral counterpart obtained after CID could be an already cyclized form of the enyne bound to gold.
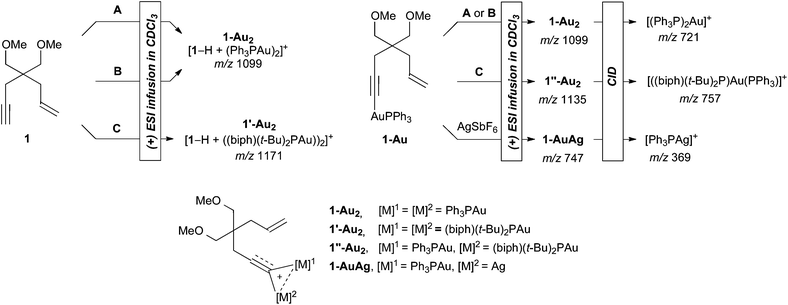 | ||
| Scheme 3 The identification of dinuclear species (biph = biphenyl-2-yl, CID = Collision Induced Dissociation). | ||
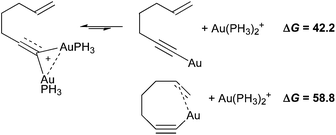 | ||
| Scheme 4 Calculated free energies of dissociation (kcal mol−1). | ||
This preliminary observation was intriguing. As mentioned above, gold acetylides and the corresponding diaurated species have been postulated as putative intermediates by Toste6 and by Gagosz,7 respectively, in the cycloisomerization of 1,5-allenynes and diynes. Although ESI spectrometry showed that such species could indeed be obtained, the transfer of this information to a solution phase catalytic event should be made with care. This drove us to the examination of the solution reactivity by NMR spectrometry. Monitoring the reaction of 1 with catalysts A or C could not be achieved due to their high activity, even at low temperatures. On the other hand, catalyst B requires heat to be active (see Table 1). At RT, using 0.33 equiv. of B, diagnostic 1H NMR signals of the free enyne, such as the propargylic protons at 2.16 ppm, were still clearly present after 5 min (Scheme 5). A new compound was observed in the 31P spectra, displaying a peak at 38.4 ppm. In 1H NMR, the propargylic protons were downfield shifted to 2.68 ppm, whereas the ethylenic ones showed virtually no changes. A broad peak at 1.64 ppm was also clearly visible. The addition of D2O to the mixture caused the disappearance of this signal, suggesting complexed water (see the supporting information for details†). However, none of 1H and 31P peaks correspond to those of the independently prepared dinuclear species 1-Au22BF44, the propargylic protons of this complex resonating at 2.80 ppm and the phosphorus atoms at 37.1 ppm. We envisaged the formation of the trinuclear dimer 7 at RT, in which two gold acetylide moieties share the same Ph3PAu+ unit. This hypothesis could be supported by a new mass spectrometry experiment: using a low cone voltage (leading to a low internal energy), the fragile species 7 was clearly observed in the ESI spectra at m/z 1739 (see the supporting information for details, Figs S4–S5†). Presumably, aggregate 7 corresponds to a stabilized form of 1-Au22BF44 in solution and not a cycloisomerization intermediate. Species 7 can be observed in mass spectrometry only at extremely low energy conditions because of its instability in the gas phase in contrast to 1-Au22BF44.
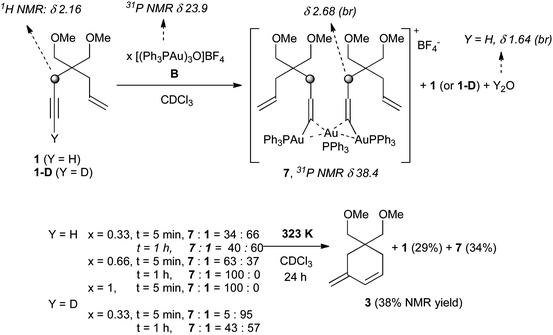
Thus, upon the addition of 0.33 equiv. of B, if our assignment is correct, a 36:64 ratio between 7 and 1 can be estimated from 1H NMR. It changed to 40:60 after 1 h. Increasing the amount of B to 0.66 equiv. gave rise to a 63:37 mixture of 7 and 1 after 5 min and only 7 could be observed after 1 h. With 1 equiv. of B, 1 was already no longer observed after 5 min. The expected kinetic isotope effect associated with the deprotonation of 1 could be evidenced by using 1-D, leading to a 5:95 mixture of 7 and 1 after 5 min using 0.33 equiv. of B. Lastly, the 37:63 mixture obtained after 1 h using 1 and 0.33 equiv. of B was heated to 50 °C for 24 h. Although the amount of 7 remained almost the same (34% vs. 37%), the NMR yield of 1 decreased to 29%, while 38% of the cycloisomerization product 3 was formed (see ESI†). Therefore, this experiment suggests that the gold acetylides derived from enynes are actually not cyclization intermediates. To gain further insights, some gold acetylides were prepared and their reactivity studied by NMR experiments. A first follow-up confirmed the formation of new complexes when the gold acetylides were mixed with a stoichiometric amount of A at RT in CDCl3 (Scheme 6, eqn (1)).
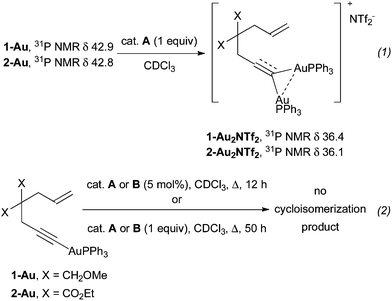 | ||
| Scheme 6 The reactivity of gold acetylide derivatives. | ||
According to the 1H, 13C and 31P NMR chemical shifts, the expected dinuclear complexes 1-Au22NTf22 and 2-Au22NTf22 were actually formed. However, when 1-Au and 2-Au were submitted to catalysts A or B (5 or 100 mol%) under refluxing conditions, no cycloisomerization product was observed, even after 50 h (eqn (2)). Thus, it seems unlikely that gold acetylides themselves may be involved in a catalytic cyclizing pathway. However, based on the mechanism proposed by Houk and Toste for 1,5-allenynes (Scheme 2),6 it can be argued that in order to have an effective catalytic cycle, a proton source is required, e.g. the enyne itself. Thus, when a mixture of 1 and 8-D (or 2 and 8-D) was engaged, no scrambling of the deuterium was observed (Scheme 7). So, it is most likely that deprotonation does not even take place in these reactions.
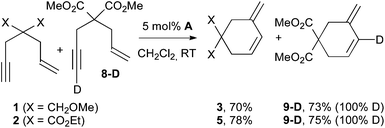 | ||
| Scheme 7 The deuterium scrambling experiment. | ||
Once we established that the starting enyne cannot act as a proton donor, we wanted to compare in solution the reactivity of the enyneversus its corresponding acetylide. Thus, we ran the following set of experiments with both substrates 1 and 2 (Table 2). Enyne 1 (or 2) was first mixed with 5 mol% of 1-Au (or 2-Au), and exposed to 5 mol% of A. Curiously, this resulted in the formation of 3 (or 5) as traces (entries 1 and 3), as if the catalysis process was inhibited. So, we decided to increase the quantity of catalyst and, very interestingly, when 5 mol% of A was further added, a clean and rapid formation of product 3 (or 5) was observed (entries 2 and 4). This series of experiments demonstrates the greater affinity of A for 1-Au (or 2-Au) than for the free enyne and further discredits the intermediacy of gold acetylides, at least at RT.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Gold acetylide | Solvent | A (mol%) | % yield |
| 1 | 1 | 1-Au | CH2Cl2 | 5 | trace |
| 2 | 1 | 1-Au | CH2Cl2 | 10 | 70 |
| 3 | 2 | 2-Au | CDCl3 | 5 | trace |
| 4 | 2 | 2-Au | CDCl3 | 10 | 70 |
Based on some findings from a related study20 and recent literature reports by Nolan,21 who showed that diaurated hydroxide (IPrAu)2OH+ can serve as a reservoir of IPrAu+ or as the active species itself in some gold-catalyzed reactions, we wondered whether (LAu)2OH+ could be involved in the formation of diaurated enyne.
Preliminary confirmation came when enyne 1 was introduced in ESI with catalyst C. An ion at m/z 1189 [1 + ((biphenyl-2-yl)(t-Bu)2PAu)2OH]+ was also observed (see the supporting information, Fig. S1†). This ion was shifted to m/z 1190 using 1-D instead of 1, whereas the m/z 1171 ion [1–H(D) + ((biphenyl-2-yl)(t-Bu)2PAu)2]+ was unchanged. Although the formation of the (IPrAu)2OH+ complex can be controlled by “proton-catalyzed” hydrolysis of IPrAu+, it could arise in our case from adventitious water and a proton source when catalyst A or C is used, or just with a proton source with catalyst B. Assuming that catalysts A and B contain traces of acid, a mixture of enyne 1 and catalyst B in CH3CN (instead of CDCl3 to allow water miscibility) was re-injected into the mass spectrometer. Using this inadequate solvent for cycloisomerizations,22 the peak at m/z 1099 ([1–H + (Ph3PAu)2]+) was now observed with a low relative abundance. Nevertheless, under these conditions, the addition of 5% of water increased the intensity of the m/z 1099 ion by a factor of 10 (see supporting information, Fig. S6†). Moreover, a new peak at m/z 1117, potentially corresponding to [1 + (Ph3PAu)2OH]+ was now weakly observed. Conversely, when enyne 1 was treated with 0.33 equiv. of B in strictly anhydrous CDCl3, formation of 7 appeared to be slower. After 5 min, the 7:1 ratio was 23:77, compared to 34:66 (Scheme 5).
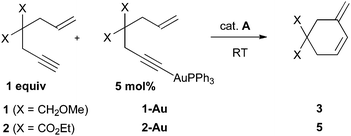
Calculations were carried out to shed more light on that matter (Scheme 8). While the formation of (H3PAu)2OH+ by hydration of H3PAu+ would be appreciably exothermic (eqn (1)), it would be quite endothermic when starting from (H3PAu)3O+ (eqn (2)). On the other hand, under acidic conditions, a strongly exothermic process may take place (eqn (3)).
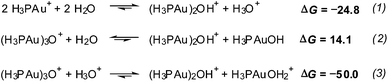 | ||
| Scheme 8 Calculated free energies for the formation of (H3PAu)2OH+. | ||
We next compared the various routes to the diaurated gold species, starting either from H3PAu+, (H3PAu)3O+, or (H3PAu)2OH+ (Scheme 9). With the former, water was chosen as a base (eqn (1)). While the complexation of H3PAu+ is strongly exothermic, the formation of the gold acetylide is hampered by a strong endothermicity. Nevertheless, the experimentally observed affinity of H3PAu+ for the gold acetylide was well reproduced computationally and the overall process is exothermic by 35.1 kcal mol−1. Starting from (H3PAu)3O+, although the liberation of H3PAu+ to complex the triple bond seems difficult to achieve (endothermic by 42.3 kcal mol−1), the formation of the gold acetylide is only slightly endothermic (eqn (2)). However, the acetylide is found to be more stable by 0.4 kcal mol−1 than the corresponding diaurated species. These results are very close to those reported by Toste and Houk in the 1,5-allenyne series,6 including the fact that the dinuclear complex is not the ground state in the gas phase. From (H3PAu)2OH+, both the dissociation/triple bond complexation and acetylide formation are quite endothermic, yet the formation of the diaurated species is exothermic by 10.3 kcal mol−1 (eqn (3)). These computations suggest that when an oxonium catalyst such as B is used, the formation of dinuclear species from enynes or allenynes could in fact involve (Ph3PAu)2OH+. This feature does not seem to be a requirement with a type Acatalyst.
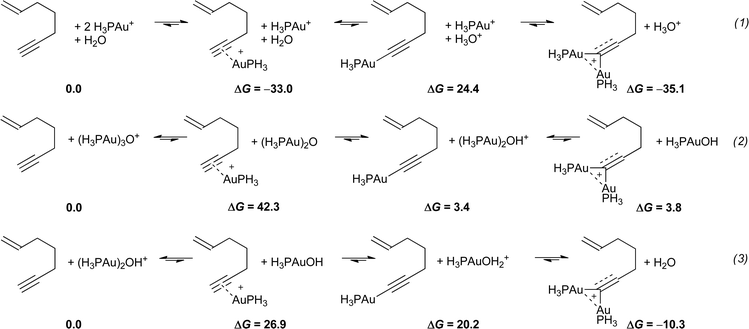 | ||
| Scheme 9 Computational prediction of the stability of the gold complexes of an enyne (free energies relative to 0.0 in kcal mol−1). | ||
In conclusion, our study confirms the very high affinity of gold acetylides for a second complexation of gold. Both types of species appear to be non-reactive in the 1,6-enyne cycloisomerization catalytic cycle. We are now working on allenyne substrates to gain more insight into the reactivity of the corresponding gold acetylide intermediates. In addition, we have observed the presence of complexes involving organic substrates with a diaurated gold hydroxide generated from the catalyst under certain conditions, which seems to confirm the role of this recently described active form of gold.
Acknowledgements
The authors thank UPMC, CNRS, ANR BLANC Allenes, Ministère de l'Enseignement Supérieur et de la Recherche (Ph. D. grant for A. S.) and IUF (MM and LF) for financial support. Calculations were performed at the CRIHAN, project 2006-13.Notes and references
- For some selected reviews, see:
(a) Z. Li, C. Brouwer and C. He, Chem. Rev., 2008, 108, 3239 CrossRef CAS
; (b) A. Arcadi, Chem. Rev., 2008, 108, 3266 CrossRef CAS
- One of the most emblematic issues perhaps being the cation vs.carbene rendition of gold species:
(a) G. Seidel, R. Mynott and A. Fürstner, Angew. Chem., Int. Ed., 2009, 48, 2510 CrossRef CAS
-
(a) A. S. K. Hashmi, Angew. Chem., Int. Ed., 2010, 49, 5232 CrossRef CAS
-
(a) M. R. Gagné and D. Weber, Org. Lett., 2009, 11, 4962 CrossRef CAS
-
(a) T. N. Hooper, M. Green and C. A. Russel, Chem. Commun., 2010, 46, 2313 RSC
- P. H.-Y. Cheong, P. Morganelli, M. R. Luzung, K. N. Houk and F. D. Toste, J. Am. Chem. Soc., 2008, 130, 4517 CrossRef CAS
- Y. Odabachian, X.-F. Le Goff and F. Gagosz, Chem.–Eur. J., 2009, 15, 8966 CrossRef CAS
- For reviews, see:
(a) V. Michelet, P. Y. Toullec and J.-P. Genêt, Angew. Chem., Int. Ed., 2008, 47, 4268 CrossRef CAS
- For relevant reports, see:
(a) C. Nieto-Oberhuber, S. Lopez, M. P. Muñoz, D. J. Cárdenas, E. Buñuel, C. Nevado and A. M. Echavarren, Angew. Chem., Int. Ed., 2005, 44, 6146 CrossRef CAS
- S. Baumgarten, D. Lesage, V. Gandon, J.-P. Goddard, M. Malacria, J.-C. Tabet, Y. Gimbert and L. Fensterbank, ChemCatChem, 2009, 1, 138 Search PubMed
- N. Mézailles, L. Ricard and F. Gagosz, Org. Lett., 2005, 7, 4133 CrossRef
-
(a) A. N. Nesmeyanov, E. G. Perevalova, Y. T. Struchkov, M. Y. Antipin, K. I. Grandberg and V. P. Dyadchenko, J. Organomet. Chem., 1980, 201, 343 CrossRef CAS
-
(a) C. Nieto-Oberhuber, S. Lopez and A. M. Echavarren, J. Am. Chem. Soc., 2005, 127, 6178 CrossRef CAS
- N. Cabello, E. Jiménez-Núñez, E. Buñuel, D. J. Cárdenas and A. M. Echavarren, Eur. J. Org. Chem., 2007, 4217 CrossRef CAS
- R. Thota, D. Lesage, Y. Gimbert, L. Giordano, S. Humbel, A. Milet, G. Buono and J.-C. Tabet, Organometallics, 2009, 28, 2735 CrossRef CAS
- It is worthy of note that, when using 1–D, an analog of 1 bearing a deuterium atom at the triple bond, with B, m/z 1099 ion was also observed.
- The structure of 1-Au was ascertained by means of a X-ray diffraction study (CCDC 794624†).
- For a recent report on the preparation of gold acetylides, see: G. C. Fortman, A. Poater, J. W. Levell, S. Gaillard, A. M. Z. Slawin, I. D. W. Samuel, L. Cavallo and S. P. Nolan, Dalton Trans., 2010, 39, 10382 Search PubMed
- Interestingly, a molecular ion is absent in the ESI mass spectrum of 1-Au alone, which rather exhibits m/z 1099. While this may question the fact that A or B actually transfer Ph3PAu+ to 1-Au in the ESI source, the results obtained with C or AgSbF6 leave no doubt about the proposed intermolecular process. When the latter two are used, m/z 1099 is still observed, but weakly.
- A. Simonneau, F. Jaroschik, D. Lesage, M. Karanik, R. Guillot, M. Malacria, J.-C. Tabet, J.-P. Goddard, L. Fensterbank, V. Gandon, Y. Gimbert to be published.
- For mono- and diaurated gold hydroxides, see:
(a) S. Gaillard, A. M. Z. Slawin and S. P. Nolan, Chem. Commun., 2010, 46, 2742 RSC
- The use of acetonitrile instead of CDCl3 results in very low conversion of 1 or 2 into 3 or 5. This is presumably due to the coordination of the solvent to gold.
Footnote |
| † Electronic supplementary information (ESI) available: Procedure for the synthesis of gold acetylides, preparation of the diaurated complexes, NMR and mass studies, computational details. CCDC reference number 794624. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00478f |
| This journal is © The Royal Society of Chemistry 2011 |