A robust and scalable synthesis of the potent neuroprotective agent (−)-huperzine A†
Maung Kyaw Moe
Tun
,
Daniel-Joachim
Wüstmann
and
Seth B.
Herzon
*
Department of Chemistry, Yale University, New Haven, Connecticut, USA. E-mail: seth.herzon@yale.edu; Fax: +1-203-432-6144; Tel: +1-203-436-8571
First published on 25th August 2011
Abstract
(−)-Huperzine A (1) is a tricyclic alkaloid that is produced in low yield by the Chinese herb Huperzia serrata. There is intense contemporary interest in clinical application of (−)-huperzine A (1) for treating neurodegenerative diseases and protecting against the lethal effects of chemical warfare agents, such as sarin and VX. We report a robust, scalable, and efficient synthesis of (−)-huperzine A (1) from (R)-4-methyl-cyclohex-2-ene-1-one (5). Our route proceeds in 35–45% overall yield, delivers (−)-huperzine A (1) in only eight steps from cyclohexenone 5, requires only three chromatographic purifications, and can provide gram quantities of the target. This route represents a critical, enabling advance toward detailed evaluation of (−)-huperzine A (1) in clinical settings.
(−)-Huperzine A (1) is a tricyclic alkaloid produced by the Chinese herb Huperzia serrata.1 (−)-Huperzine A (1) is a potent, selective, and reversible inhibitor of acetylcholine esterase (AChE, Ki = 23 nM).2 Recent studies have established that this activity may be exploited to counteract organophosphate chemical warfare agents, such as sarin and VX, by inhibiting their covalent modification of peripheral and cerebral AChE.3 A large body of evidence also suggests that (−)-huperzine A (1) may slow the progression of neurodegenerative diseases, including Alzheimer's disease.4 (−)-Huperzine A (1) is well-tolerated in humans, even at doses well above those required clinically.5 Consequently, clinical investigation of (−)-huperzine A (1) is a subject of intense research in the pharmaceutical and defense industries.
The primary obstacle to the clinical development of (−)-huperzine A (1) has been one of supply. Extraction from natural sources is low-yielding (average yield = 0.011% from the dried herb),4a and overharvesting has caused a rapid decline in the abundance of Huperziaceae.6 Compounding these issues, the producing species requires nearly 20 years to reach maturity.6
Total synthesis offers an alternative potential source of huperzine. An enantioselective synthesis is highly desirable, because (+)-huperzine A is significantly less potent than the natural (−)-antipode (1).7 The first total syntheses of (±)-huperzine A were reported by Kozikowski and Xia8 and Qian and Ji.9 A chiral auxiliary-based route was later developed by Kozikowski et al.10 In the interceding years, several research groups have reported modifications to the Kozikowski route,11 as well as complete,12 partial,13 and formal14 routes to huperzine. Nonetheless, Kozikowski's chiral controller-based route,10 which proceeds in 16 steps and ca. 2.8% yield, remains the most efficient published pathway to synthetic (−)-huperzine A (1).15
The majority of approaches to huperzine A have relied on introduction of a four-carbon fragment to a bicyclic structure (retrosynthetic cleavage of bonds a and b in 1, Scheme 1). We envisioned a distinct approach, wherein disconnection of two alternative bonds (see 2) forms the ketone and pyridone-based synthons 3 and 4, respectively. The former might be obtained from (R)-4-methyl-cyclohex-2-ene-1-one (5) while 3-bromo-2-(bromomethyl)-6-methoxypyridine (6) would serve as a functional equivalent to 4. The C-4 stereocenter in 5 is used in our route to control relative and absolute stereochemistry in the target. Several convenient methods to prepare (R)-4-methyl-cyclohex-2-ene-1-one (5) have been reported.16 We elected to use a straightforward four-step sequence starting from (+)-pulegone.16a Dihalopyridines such as 6 have found use in a distinct and significantly more lengthy route to (−)-huperzine A (1),14c as well as in the synthesis of other Lycopodium alkaloids.17
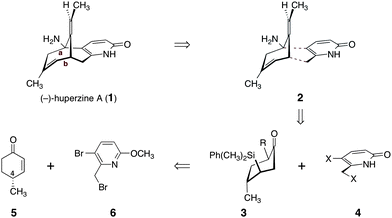 | ||
| Scheme 1 Retrosynthetic analysis of (−)-huperzine A (1). | ||
The successful implementation of this strategy is shown in Scheme 2. To render the route amenable to large-scale synthesis, we extensively optimized each step, and this allowed many transformations to be efficiently telescoped (the final synthetic route requires three chromatographic purification steps). Our work commenced with conjugate addition of lithium dimethylphenylsilylcuprate to (R)-4-methyl-cyclohex-2-ene-1-one (5). Alkylation of the incipient enolate with 3-bromo-2-(bromomethyl)-6-methoxypyridine (6) afforded the addition–alkylation product 7 as a single detectable diastereomer (1H NMR analysis), isolated in 84–91% yield after purification (2.0–4.5 g scale). Kinetically-controlled deprotonation of 7 and trapping of the resulting enolate with para-toluenesulfonyl cyanide,18 followed by immediate work up of the product mixture, formed the α-cyanoketone 8 in high purity (est. >95%, 1H NMR analysis). Rapid isolation of the product was critical, as the α-cyanoketone 8 underwent disproportionation to starting material (7) and an α,α-dicyanoketone (not shown) if the mixture was allowed to age.
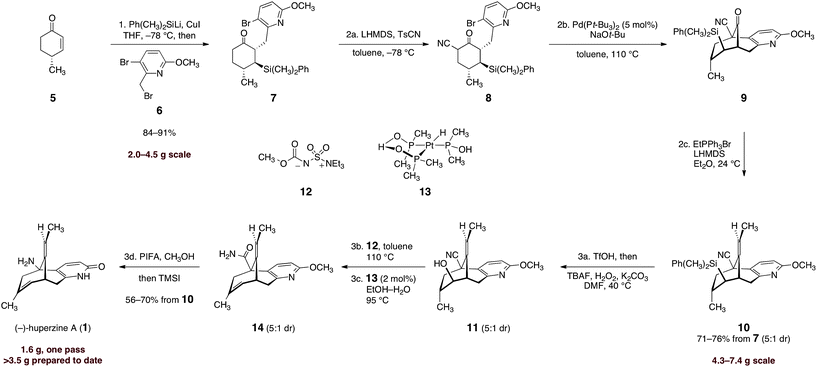 | ||
Scheme 2 Enantioselective synthesis of (−)-huperzine A (1). Reagents and conditions: (1) Ph(CH3)2SiLi, CuI, HMPA, THF, −78 → −23 → −78 °C, then 6, −78 → −23 °C, 84–91%; (2a) LHMDS, p-TsCN, toluene, −78 °C; (2b) Pd(Pt-Bu3)2 (5 mol%), NaOt-Bu, toluene, 110 °C; (2c) EtPPh3Br, LHMDS, Et2O, 24 °C, 71–76% from 7, E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Z = 5:1; (3a) TfOH, DCM, 0 → 24 °C, then TBAF, H2O2, K2CO3, DMF, 40 °C, E:Z = 5:1; (3b) 12, toluene, 110 °C, E:Z = 5:1; (3c) 13 (2 mol%), EtOH–H2O (2:1), 95 °C, E:Z = 5:1; (3d) PIFA, CH3OH, reflux, then TMSI, CHCl3, reflux, then CH3OH, reflux, 56–70% from 10. :Z = 5:1; (3a) TfOH, DCM, 0 → 24 °C, then TBAF, H2O2, K2CO3, DMF, 40 °C, E:Z = 5:1; (3b) 12, toluene, 110 °C, E:Z = 5:1; (3c) 13 (2 mol%), EtOH–H2O (2:1), 95 °C, E:Z = 5:1; (3d) PIFA, CH3OH, reflux, then TMSI, CHCl3, reflux, then CH3OH, reflux, 56–70% from 10. | ||
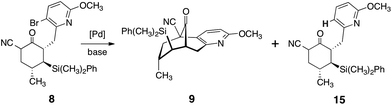
The unpurified α-cyanoketone 8 was then subjected to a palladium-catalyzed intramolecular enolate heteroarylation.19 Among several catalyst precursors examined, bis(tri-tert-butylphosphine)palladium (0), prepared by the method of Dai and Fu,20 emerged as the most effective. A dramatic dependence on base was observed (Table 1). Thus, in the presence of carbonate bases (entries 1, 2), the protodebrominated product 15 predominated. Sodium hydride (entry 3) improved conversion to the cyclized product (9), although extensive decomposition also occurred. Ultimately, we identified sodium tert-butoxide (entry 5) as optimal, and using this base the product was obtained in essentially quantitative yield (1H NMR analysis). The next step of the sequence called for the stereoselective olefination of the ketone function of 9. Treatment of 9 with the lithium ylide derived from ethyltriphenylphosphonium bromide (ether, 24 °C) afforded the olefination product 10 in high yield. A clear trend between E:Z selectivity and concentration was observed (E/Z = 1.1, 1.8, 5 at 1.0, 0.1, and 0.01 M, respectively), which is consistent with a salt effect and suggests the desired E-isomer is the kinetically-favored product.21 Under optimized conditions, the olefinated product 10 was isolated in 71–76% yield from 7 as a 5:1 mixture of E/Z isomers by flash-column chromatography (4.3–7.4 g scale). By this approach, the entire carbon skeleton of 1 was formed in high overall yield and in four steps on a multi-gram scale.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Base | mol% Pd | Yield 9b | Yield 15c | Dec.c |
| a All reactions were conducted using Pd(Pt-Bu3)2 as precatalyst in toluene at 110 °C for 3 h. b Isolated yield after purification by flash-column chromatography. c Estimated by 1H NMR and LC/MS analysis of the unpurified reaction mixture. Dec. = decomposition. | |||||
| 1 | K2CO3 | 10 | <1% | 99% | — |
| 2 | Na2CO3 | 10 | <1% | 99% | — |
| 3 | NaH | 10 | 50% | <1% | 30% |
| 4 | KOt-Bu | 10 | 64% | 10% | 20% |
| 5 | NaOt-Bu | 5 | >99% | <1% | <1% |
Treatment of the olefination product (10) with trifluoromethanesulfonic acid, followed by oxidative desilylation, provided the cyanoalcohol 11 in high purity (1H NMR analysis). The unpurified cyanoalcohol 11 was efficiently dehydrated by heating with the Burgess reagent (12) in toluene. Thermolysis of the dehydrated product (not shown) in the presence of the platinum catalyst 1322 in aqueous ethanol afforded the amide 14. Finally, Hofmann rearrangement [bis(trifluoroacetoxy)iodobenzene], global deprotection, and purification by flash-column chromatography afforded separately (−)-huperzine A (1, 56–70% over four operations) and its olefin isomer (not shown, 11–14%). Synthetic (−)-huperzine A (1) was identical in all respects (1H NMR, 13C NMR, IR, three TLC solvent systems, LC/MS retention time, optical rotation) to an authentic sample. Batches of (−)-1 as large as 1.6 g have been prepared.23
To date, over 3.5 g of (−)-huperzine A (1) have been prepared by the route delineated above. Our synthesis proceeds in 35–45% overall yield (16-fold more efficient than any other previously reported enantioselective route), and requires only three chromatographic purifications. We envision that this chemistry will provide a reliable supply of synthetic (−)-huperzine A (1) and will greatly facilitate its clinical development for neuroprotective and anti-neurodegenerative applications.
Notes and references
- (a) J.-S. Liu, Y.-L. Zhu, C.-M. Yu, Y.-Z. Zhou, Y.-T. Han, F.-W. Wu and B.-F. Qi, Can. J. Chem., 1986, 64, 837 CrossRef CAS. Selected review: (b) A. P. Kozikowski and W. Tückmantel, Acc. Chem. Res., 1999, 32, 641 CrossRef CAS.
- (a) Y. E. Wang, D. X. Yue and X. C. Tang, Acta. Pharmacol. Sin., 1986, 7, 110 CAS. For the structure of (−)-huperzine A (1) bound to AChE, see: (b) M. L. Raves, M. Harel, Y.-P. Pang, I. Silman, A. P. Kozikowski and J. L. Sussman, Nat. Struct. Biol., 1997, 4, 57 CrossRef CAS.
- (a) G. Lallement, J.-P. Demoncheaux, A. Foquin, D. Baubichon, M. Galonnier, D. Clarençon and F. Dorandeu, Drug Chem. Toxicol., 2002, 25, 309 CrossRef CAS; (b) R. Gordon, J. Haigh, G. Garcia, S. Feaster, M. Riel, D. Lenz, P. Aisen and B. Doctor, Chem.-Biol. Interact., 2005, 157–158, 239 CrossRef CAS; (c) J. Haigh, S. Johnston, A. Peppernay, P. Mattern, G. Garcia, B. Doctor, R. Gordon and P. Aisen, Chem.-Biol. Interact., 2008, 175, 380 CrossRef CAS; (d) J. Z. Karasova, J. Bajgar, L. Novotny and K. Kuca, Lett. Drug Des. Discovery, 2009, 6, 563 CrossRef CAS. For a review, see: (e) G. Lallement, V. Baille, D. Baubichon, P. Carpentier, J.-M. Collombet, P. Filliat, A. Foquin, E. Four, C. Masqueliez, G. Testylier, L. Tonduli and F. Dorandeu, NeuroToxicology, 2002, 23, 1 CrossRef CAS.
- For selected reviews, see: (a) D. L. Bai, X. C. Tang and X. C. He, Curr. Med. Chem., 2000, 7, 355 CAS; (b) R. Wang, H. Yan and X.-c. Tang, Acta Pharmacol. Sin., 2006, 27, 1 CrossRef CAS; (c) H. Y. Zhang and X. C. Tang, Trends Pharmacol. Sci., 2006, 27, 619 CrossRef CAS; (d) H. Y. Zhang, C. Y. Zheng, H. Yan, Z. F. Wang, L. L. Tang, X. Gao and X. C. Tang, Chem.-Biol. Interact., 2008, 175, 396 CrossRef CAS.
- J. T. Little, S. Walsh and P. S. Aisen, Expert Opin. Invest. Drugs, 2008, 17, 209 CrossRef CAS.
- X. Ma, C. Tan, D. Zhu and D. R. Gang, J. Ethnopharmacol., 2006, 104, 54 CrossRef CAS.
- T. Xi-Can, G. H. Kindel, A. P. Kozikowski and I. Hanin, J. Ethnopharmacol., 1994, 44, 147 CrossRef.
- Y. Xia and A. P. Kozikowski, J. Am. Chem. Soc., 1989, 111, 4116 CrossRef CAS.
- L. Qian and R. Ji, Tetrahedron Lett., 1989, 30, 2089 CrossRef CAS.
- F. Yamada, A. P. Kozikowski, E. R. Reddy, Y. P. Pang, J. H. Miller and M. McKinney, J. Am. Chem. Soc., 1991, 113, 4695 CrossRef CAS.
- (a) S. Kaneko, T. Yoshino, T. Katoh and S. Terashima, Heterocycles, 1997, 46, 27 CrossRef CAS; (b) S. Kaneko, T. Yoshino, T. Katoh and S. Terashima, Tetrahedron: Asymmetry, 1997, 8, 829 CrossRef CAS; (c) S. Kaneko, T. Yoshino, T. Katoh and S. Terashima, Tetrahedron, 1998, 54, 5471 CrossRef CAS; (d) C. Chassaing, A. Haudrechy and Y. Langlois, Tetrahedron Lett., 1999, 40, 8805 CrossRef CAS; (e) X.-C. He, B. Wang, G. Yu and D. Bai, Tetrahedron: Asymmetry, 2001, 12, 3213 CrossRef CAS; (f) Q.-B. Pan and D.-W. Ma, Chin. J. Chem., 2003, 21, 793 CrossRef CAS.
- T. Koshiba, S. Yokoshima and T. Fukuyama, Org. Lett., 2009, 11, 5354 CrossRef CAS.
- J. Ward and V. Caprio, Tetrahedron Lett., 2006, 47, 553 CrossRef CAS.
- (a) A. Haudrechy, C. Chassaing, C. Riche and Y. Langlois, Tetrahedron, 2000, 56, 3181 CrossRef CAS; (b) I. Y. C. Lee, M. H. Jung, H. W. Lee and J. Y. Yang, Tetrahedron Lett., 2002, 43, 2407 CrossRef CAS; (c) C. Lucey, S. A. Kelly and J. Mann, Org. Biomol. Chem., 2007, 5, 301 RSC.
- The most efficient route to racemic huperzine proceeds in 12 steps and 8.9% overall yield. See, ref. 8 and: A. P. Kozikowski, E. R. Reddy and C. P. Miller, J. Chem. Soc., Perkin Trans. 1, 1990, 195 RSC.
- (a) H. W. Lee, S. K. Ji, I.-Y. C. Lee and J. H. Lee, J. Org. Chem., 1996, 61, 2542 CrossRef CAS; (b) F. Bertozzi, P. Crotti, B. L. Feringa, F. Macchia and M. Pineschi, Synthesis, 2001, 483 CrossRef CAS; (c) R. Naasz, L. A. Arnold, A. J. Minnaard and B. L. Feringa, Angew. Chem., Int. Ed., 2001, 40, 927 CrossRef CAS.
- V. Bisai and R. Sarpong, Org. Lett., 2010, 12, 2551 CrossRef CAS.
- D. Kahne and D. B. Collum, Tetrahedron Lett., 1981, 22, 5011 CrossRef CAS.
- (a) M. Kawatsura and J. F. Hartwig, J. Am. Chem. Soc., 1999, 121, 1473 CrossRef CAS; (b) J. M. Fox, X. Huang, A. Chieffi and S. L. Buchwald, J. Am. Chem. Soc., 2000, 122, 1360 CrossRef CAS. For a review, see: (c) F. Bellina and R. Rossi, Chem. Rev., 2010, 110, 1082 CrossRef CAS.
- C. Dai and G. C. Fu, J. Am. Chem. Soc., 2001, 123, 2719 CrossRef CAS.
- See A. B. Reitz, S. O. Nortey, A. D. Jordan, Jr., M. S. Mutter and B. E. Maryanoff, J. Org. Chem., 1986, 51, 3302 and references therein Search PubMed.
- T. Ghaffar and A. W. Parkins, J. Mol. Catal. A: Chem., 2000, 160, 249 CrossRef CAS.
- As a consequence of the potent activity and high skin permeability of 1, we have not attempted to prepare larger batches in our laboratory.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and detailed characterization data of all new compounds. See DOI: 10.1039/c1sc00455g |
| This journal is © The Royal Society of Chemistry 2011 |