Foldamer with a spiral perylene bisimide staircase aggregate structure†
Volker
Dehm
a,
Michael
Büchner
a,
Joachim
Seibt
b,
Volker
Engel
b and
Frank
Würthner
*a
aUniversität Würzburg, Institut für Organische Chemie and Center for Nanosystems Chemistry, Am Hubland, 97074, Würzburg, Germany. E-mail: wuerthner@chemie.uni-wuerzburg.de
bUniversität Würzburg, Institut für Physikalische und Theoretische Chemie, Am Hubland, 97074, Würzburg, Germany
First published on 30th August 2011
Abstract
Although the precise control of dye–dye interactions is known to be of fundamental importance to achieve desired functional properties in π-conjugated materials, there are only sporadic examples where precise structural control of dye–dye interactions could be achieved by the attachment of the chromophores to an oligomeric or polymeric backbone. In this edge article, we introduce an oligophenylene ethynylene/perylene bisimide (PBI) dye system which is conceived by the foldamer concept. The molecular design enables folding of the π-conjugated oligomer to give a geometrically well-defined π-stack of intrastrand PBI molecules. The synthesis of the foldamer and its characterization by MALDI-TOF mass spectrometry are described, and the average size is estimated by gel permeation chromatography (GPC) and diffusion ordered NMR spectroscopy (DOSY). The folding dynamics are demonstrated by ultraviolet/visible absorption and fluorescence emission spectroscopic studies as the folded and unfolded states possess distinct optical properties. Quantum dynamic considerations revealed a good agreement of the calculated aggregate absorption spectrum with those obtained experimentally.
Introduction
Nature often utilizes rigid scaffolds to organize π-conjugated molecules into highly ordered structures, creating biomacromolecules of pivotal functions. A splendid example is given by the self-ordering of DNA into double helices,1 which serve as a unique storage system for the genetic information. Likewise, many natural light harvesting (LH) systems, e.g., the LH system of purple bacteria,2 rely on rigidifying protein frameworks to organize chlorophyll dyes into efficient LH complexes. The past decades have witnessed enormous efforts to mimic such functional biological structures. However, even the most promising imitations so far have failed to be genuinely competitive with natural archetypes. The improvement of functional properties of artificial dye assemblies, for instance, has often relied until now more or less on intuitive “trial-and-error”, a time-consuming approach, which rarely leads to the best conceivable results. A more purposeful approach should be the elaboration of a profound understanding of the dye–dye interactions, providing rational structure–property relationships. This would open the door to develop more general concepts for directed and efficient synthesis of artificial dye assemblies with desired properties. However, towards this goal suitable model systems with clearly defined structures are needed that facilitate the investigation of electronic interactions between chromophore molecules.One of the most promising strategies to generate synthetic macromolecules with orientational control of attached functional building blocks is provided by the foldamer concept. Systems that are capable of folding into defined structures have attracted increasing advertence during the past decade,3 prominent examples being the oligophenylene ethynylene foldamers reported by Moore et al.4 and the aromatic oligoamides of the Huc group.5 More recently, the first examples of foldamer systems with photofunctional properties, i.e. circular dichroism based chirality sensing, photoswitchable folding and folding-dependent photoinduced electron transfer, have been reported.6
One of the most interesting features of foldamers is their unique conformational ordering in the folded state, the structure of which can often be predicted, e.g., by performing rather simple molecular modeling studies. However, the fusion of rigid foldable backbones driven by the strong π–π interactions between dyes to generate highly ordered aggregates with predictable geometries by folding is currently unprecedented.
We are particularly interested in controlling the organization of perylene bisimide (PBI) dyes, a class of lightfast and chemically robust chromophores with intriguing optical as well as material properties.7 Owing to their outstanding n-type semiconducting properties, PBI dyes have been applied in organic electronic devices such as organic solar cells, organic light emitting diodes and field effect transistors.8 However, the control of their functional properties is still dominated by serendipity as these dyes may pack in quite different geometrical arrangements in the solid state.7 To reveal structure–property relationships in PBI π-stacks we and others were able within the past years to synthesize several examples of covalently linked PBI dimers and trimers with well-defined geometries.9 Due to their small size, however, material properties of extended π stacks cannot easily be derived from such model systems. On the other hand, many interesting covalent PBI oligomers10 and polymers11 with a dense packing of the chromophores have been reported recently but, unfortunately, their structures could be determined only on a vague scale, and their proposed models cannot be taken for granted when it comes to the unequivocal determination of mutual orientation of the dye monomers. For our purposes, the desired model systems are thus species of oligomeric size with clearly defined geometry in the assembled structure.12 Hence, after performing thorough geometrical analyses by molecular modeling, we have designed the oligomeric target model system 1 as depicted in Fig. 1. The conjunction of the PBI dye units is realized by a rigid oligophenylene ethynylene (OPE) framework with ortho–meta alternating connectivity, which upon folding forces the system into a spiral staircase motif. As a result, a π–π distance of 3.4 Å for the PBI dyes and a defined rotational displacement of dye molecules of approximately 50° is obtained, whereas longitudinal and transversal shifts of the dyes are absent.
 | ||
| Fig. 1 Folding of PBI oligomer 1 into helical H-type aggregates. (a) Chemical structure of oligomer 1, (b) geometry optimized structure of an unfolded state of 1, (c) side view (top) and top view (bottom) of a geometry optimized structure of the folded state, (d) and (e) top and side view of a dimeric subunit of the folded oligomer, respectively. Hydrogens and alkyl chains are omitted for clarity. | ||
Results and discussion
PBI oligomer 1 was synthesized according to the route outlined in Scheme 1. Perylene monoimide 213 was condensed with 3,5-diiodoaniline14 to afford the unsymmetrically substituted PBI 3. A subsequent Sonogashira cross-coupling co-polymerization reaction of PBI 3 with 1,2-diethynyl-4,5-dihexylbenzene15 afforded the desired PBI oligomer 1 as a crude product (synthetic details are given in the ESI†). The GPC analysis of this crude product (Fig. 2a) shows an intriguing feature: exactly at the length where the macromolecule can fold into a π-stack with one helical turn (Fig. 1) an abrupt termination of the polymerization is observed. In the next step the crude product was purified by column chromatography, followed by semi-preparative gel permeation chromatography (GPC) to remove impurities and smaller-sized oligomers of 1. From the subsequent analytic GPC analysis, a weight-average molecular mass (Mw) of 8500 Da was obtained for the oligomer 1 (Fig. 2a). Given a molecular weight for one repeating unit (i.e., AB, where A denotes the PBI and B the diethynylbenzene subunit, see inset in Fig. 2b), of approximately 1080 Da, this corresponds to an average oligomer size containing eight PBI units. From the very small index of polydisperity (D = 1.1) a sharp size distribution can be deduced. Accordingly, the amounts of lower mass oligomers (< tetramer) are negligible and the presence of molecular PBI monomers can be excluded. The average molecular dimension estimated from GPC analysis corresponds well with the results obtained from diffusion-ordered NMR spectroscopy (DOSY), the latter revealed an average size of approximately nine PBI units per oligomer. For details on the size determination of 1 based on DOSY NMR spectroscopy, see the ESI.† | ||
| Scheme 1 Synthesis of PBI oligomer 1. Reagents and conditions: (i) 3,5-diiodoaniline, imidazole, pyridine, sealed flask, argon atmosphere, 150 °C, 2 h, 73% yield of 3; (ii) 1,2-diethynyl-4,5-dihexylbenzene, (allylPdCl)2, PtBu3, CuI, HNiPr2, toluene, argon atmosphere, 70 °C, 3 d, 19% yield of 1. | ||
![(a) Analytic gel permeation chromatogram of a dilute solution of PBI 1 in THF at 20 °C, before (dashed line) and after (solid line) performing twofold semi-preparative GPC. Mw = 8490 Da; Mp = 7970 Da; D = 1.10. (b) MALDI-TOF mass spectrum of 1. Conditions: chloroform solution of 1, matrix 2-[(2E)-3-(4-tert-butylphenyl)-2-methylprop-2-enylidene] malononitrile in tetrahydrofuran.](/image/article/2011/SC/c1sc00435b/c1sc00435b-f2.gif) | ||
| Fig. 2 (a) Analytic gel permeation chromatogram of a dilute solution of PBI 1 in THF at 20 °C, before (dashed line) and after (solid line) performing twofold semi-preparative GPC. Mw = 8490 Da; Mp = 7970 Da; D = 1.10. (b) MALDI-TOF mass spectrum of 1. Conditions: chloroform solution of 1, matrix 2-[(2E)-3-(4-tert-butylphenyl)-2-methylprop-2-enylidene] malononitrile in tetrahydrofuran. | ||
The structural integrity of the oligomer 1 might suffer from side reactions, e.g., oxidative homo-coupling of two acetylene units resulting in butadiyne subunits.16 The presence of such defects would seriously affect the periodical folding motif within the oligomeric system. However, on the basis of MALDI-TOF mass spectrometric analysis (Fig. 2b and Table S1 in the ESI†), such anomalies could actually be ruled out, since all prominent mass peaks are assignable to systems following the rule [AxByH2 + z·HCl]− (x = 2, 3, …, 8; y = x − 1, x, x + 1; z = 0, 1, 2; HCl adducts originate from the CHCl3 solution used for the mass spectrometric analysis). Moreover, peaks of obvious defects, e.g., [AxBx+2H2]−, are absent. It is worth mentioning that iodinated products were not observed. Apparently, this is an outcome of the competing dehalogenation process during the cross-coupling reaction, which is a limiting factor for the size of the oligomer.
Comparison of the obtained mass values with the average molecular mass determined by GPC and DOSY NMR reveals deviations, which need to be addressed: the distinct MALDI mass spectrum shown in Fig. 2b was obtained after accumulating six individual mass spectra. Still, only lower molecular weight oligomers could be observed, whereas larger species (> octamer) are absent due to methodological restrictions. Firstly, the MALDI conditions may lead to in-source decay,17 thus lower mass species of n-mer 1 may be enriched. Secondly, the mass-dependent desorption and detection process by MALDI17a is known to cause considerable underestimation of ions with higher masses compared to the results obtained by GPC analysis.18,19
Thus, predominantly smaller species and fragments of n-mer 1 are detected in MALDI mass, leading to rather low molecular mass values obtained for n-mer 1 when compared to the values obtained from GPC and DOSY experiments. Taking these facts into consideration, the presented mass spectrum (Fig. 2b) corroborates the claimed chemical structure (see Fig. 1), in particular the absence of homo-coupling products, but is ineligible to deduce a reliable size distribution for n-mer 1.
To gain insight into the optical properties of dyen-mer 1 upon π–π aggregation solvent-dependent UV/vis spectra were recorded. Two important regions in the absorption spectra have to be considered, i.e. the UV part (< 400 nm), composed of OPE absorption but also of higher energy absorption of the PBI moiety, and the visible part (400–600 nm) which can be assigned exclusively to electronic S0–S1 transition of the PBIs. For the latter (Fig. 3), distinct dependencies on the nature of the solvent can be observed, which is indicative of the degree of aggregation (π–π-stacking) of the PBI dyes of n-mer 1 (see also Table S2 in the ESI†).
 | ||
| Fig. 3 UV/vis absorption spectra of n-mer 1 (concentration < 10 mg l−1) in various solvents of different polarities at 20 °C. For better understanding the spectra are normalized to the λmax2 absorption maximum. | ||
Strong differences in the visible spectra for n-mer 1 were observed between apolar aliphatic solvents with low polarizability (methylcyclohexane (MCH), di-n-butylether) and halogenated (e.g.chloroform) or aromatic (e.g.toluene) solvents with high polarizability. In contrast, for the UV region (< 400 nm), the only notable difference in the absorption spectra of n-mer 1 in chloroform and MCH, respectively, is a minor hypochromic shift for the latter (see Fig. S2 in the ESI†), but the shapes of the spectra are almost identical. The moderate hypochromic shift may possibly originate from specific solvent interactions of the OPE scaffold and, in addition, changes in the UV absorption characteristics of the PBIs in the aggregated and non-aggregated state. Hence, while the PBIs reveal strong indications of changes in π–π-stacking interactions upon variation of the solvent, the OPE scaffold does not. This result is in excellent agreement with our structural model which does not indicate any π–π-stacking interactions between the OPE units, i.e. a feature that clearly distinguishes our novel foldamers from those reported by the Moore group.4a,b
Temperature-dependent UV/vis absorption experiments of a dilute n-mer 1 solution in MCH (for temperature-dependent UV/vis spectra of 1 in MCH, see Fig. S3a in the ESI†) revealed no pronounced changes in the absorption spectra and the degree of aggregation of the PBIs is virtually indifferent to temperature in this particular solvent. This is in contrast to our previously reported observations on the formation of intermolecularPBI π–π self-assemblies20 of 4a,b which exhibit distinct spectral changes upon temperature variation of a concentrated (2 × 10−3 M) MCH solution (solid lines in Fig. S3b in the ESI†) and even revealed a monomer absorption spectrum at more dilute concentrations (see dashed line in Fig. S3b in the ESI†).
UV/vis studies in solvent mixtures (Fig. 4) were conducted to further explore the thermodynamic driving forces for PBI aggregation in 1 (Fig. 4a). In pure nonpolar solvent MCH, a spectrum was observed with two absorption maxima λmax1 = 524 nm and λmax2 = 491 nm. The ratio Amax1/Amax2 is known to change dramatically upon formation of π stacks of PBI dyes, which is thus consulted casually to express the degree of aggregation.9c,21 Accordingly, a Amax1/Amax2 intensity ratio of 0.88 was obtained in MCH, which resembles a band shape that is generally observed for PBI π–π aggregates. Upon increasing the fraction of chloroform from 0 up to 50 vol% (red lines in Fig. 4a), a substantial increase in intensity of both absorption bands was observed, along with a change of their intensity ratio (see Fig. 4b). From 50 vol% chloroform onwards, however, only minor changes were observed (black lines in Fig. 4a). For the final solution of n-mer 1 in pure chloroform three absorption maxima λmax1 = 530 nm, λmax2 = 493 nm and λmax3 = 464 nm can be observed. These maxima resemble the vibronic progression of the electronic S0–S1 transition of uncoupled PBI monomers; the connecting OPE scaffold within n-mer 1 allows only for a maximum center-to-center distance of adjacent PBIs of approximately 2.4 nm (24 Å), according to molecular modeling studies, which is about 7 times larger than the center-to-center distance in the π–π stacked state (3.4 Å). Therefore, the observed absorption spectrum of n-mer 1 in chloroform is still influenced by interactions between randomly oriented dyes, which is expressed in a rather broad band and an Amax1/Amax2 intensity ratio of 1.12. In contrast, for a PBI monomer solution (see dashed line in Fig. S3b in the ESI†) a corresponding band ratio of typically 1.65 is obtained. Hence, the observed spectrum in chloroform does not originate from PBI units which are completely free of dye–dye interactions. The discrepancy with a pure monomer spectrum can be explained in terms of appreciable exciton coupling of the PBI chromophores that are evoked by their close spatial adjacency, enforced by the backbone and not by π–π-stacking.
 | ||
| Fig. 4 (a) Solvent-dependent UV/vis experiments of n-mer 1 at a concentration of 4.08 mg l−1 at 20 °C, starting in pure MCH and increasing the volume fraction of chloroform in 10% steps. Arrows indicate the spectral changes upon increasing the volume fraction of chloroform. (b) Plot of the intensity ratios of the absorbance at 530 and 495 nm versussolvent composition. (c) Plot of the absorbance of n-mer 1 at 530 nm (■) and 495 nm (▼) against the vol% of chloroform in MCH. (d) Plot of αunfolded of n-mer 1 at 530 nm and 495 nm against the vol% of chloroform in MCH (calculated from eqn (2)). (e) Plot of the ΔG values for the folding process of n-mer 1 derived from the spectral development at 530 nm and 495 nm. The black and the red lines are the respective fitting results from linear regression analysis according to eqn (5).4a | ||
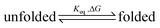
The data collected so far on the π–π aggregation of the PBI chromophores within 1 justify the conclusion that this macromolecular structure is able to reversibly switch between non-aggregated and aggregated species, i.e. a folding reaction takes place which can be biased by the nature of the solvent. For a two-state equilibrium process between an unfolded and a folded conformation (eqn (1)), we may analyze the solvent-dependent UV/vis data from Fig. 4a to determine the equilibrium constants Keq and the free energy changes ΔG for folding in the respective solvent.4a
 | (1) |
First, the absorbance at the two absorption wavelengths at 530 and 495 nm was plotted versussolvent composition, ranging from pure MCH to pure chloroform (Fig. 4c). By assuming that the respective absorbance values AF obtained in pure MCH and AU obtained in pure chloroform correspond to the respective state where either all the oligomers are folded or unfolded, one can calculate the mole fraction of unfolded species present in solution αunfolded for the remaining solvent compositions from the respective absorbance A (see Fig. 4d) according to eqn (2):
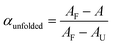 | (2) |
For each solvent composition, the equilibrium constant Keq and the related Gibbs free energy of folding ΔG can be calculated according to eqn (3) and eqn (4):
 | (3) |
| ΔG = −RTlnKeq | (4) |
| ΔG = ΔG(MCH) − m[CHCl3] | (5) |
Accordingly, the determination of the free energy change ΔG(MCH) for the pure MCH solution of n-mer 1 from the data at 495 and 530 nm, respectively, gave values of −4.2 ± 0.4 kJ mol−1 and −3.9 ± 0.2 kJ mol−1, indicating a clear preference for the folded state in MCH solution.
Since π–π-aggregated PBIs are known to exhibit excimer-like emission,20 the fluorescence spectra of n-mer 1 were of particular interest. For the dilute chloroform solution, we observed three emission bands (Fig. 5, solid line). The maxima at 533 and 577 nm resemble the typical vibronically resolved PBI monomer emission from the excited S1 state into the S0 ground state.7a In addition, a broad structureless and strongly red-shifted emission band arises at about 625 nm, which can be attributed to an excimer-type emission band.
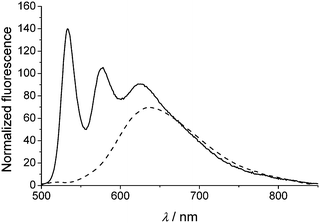 | ||
| Fig. 5 Fluorescence spectra of 1 in chloroform (solid line) and MCH solution (dashed line). The spectra are normalized to the absorbance at the excitation wavelength (λex = 485 nm), and were measured under high dilution conditions (ODmax < 0.05). | ||
The pronounced monomer emission bands confirm the presence of non π–π-stacked PBI dyes, i.e. an unfolded oligomer chain in chloroform. The occurrence of the excimer emission band can be explained due to the close proximity of adjacent PBIs (< 2.4 nm), enabling partial folding in the excited state to give excimers, or small fractions of π–π-stacked PBIs within mostly unfolded strands. This partial folding upon optical excitation is a reasonable result and pinpoints the increase of π–π-interaction energy between a PBI in the ground state and another one in the more polarizable excited state. In the less polar solvent MCH (Fig. 5, dashed line) the monomer emission band at about 520 nm has almost vanished, and a prominent excimer-like broad emission band from 550 nm up to 850 nm with a maximum at about 640 nm is obtained, indicating strong π–π-interactions of the PBI dyes in the folded state.
Recently, we have reported quantum dynamic calculations on π–π stacks of self-assembled PBI dyes 4 based on the multiconfiguration time-dependent Hartree (MCTDH) method.23 Model Hamiltonians were employed, which include a single vibrational degree of freedom for each monomer unit. As a result, good agreement of theoretically calculated aggregate spectra24 (see Fig. 6) with the ones obtained from temperature-dependent UV/vis experiments of PBI dye π–π stacks of 4a,b was demonstrated.20 Hence, for PBI π-stacks formed by self-assembly of 4a,b in solution, an aggregate geometry with a rotational displacement of 28° and a coupling, in terms of the exciton coupling theory,25 of ε = 0.065 eV could be estimated. In contrast to those self-assembled PBI aggregates, the geometry of folded n-mer 1 (with the rotational offset of the dyes being fixed to φ = 50° see Fig. 1) is controlled by the OPE scaffold and the aggregates cannot relax into the π–π-stacked state of lowest energy that is given at a rotational angle of ∼30° according to quantum chemical calculations.24
 | ||
| Fig. 6 Comparison of the optical properties of the folded PBI 1 in a dilute methylcyclohexane solution (red line) and self-assembled perylene bisimide 4a in methylcyclohexane at a concentration of 2 × 10−3 M (black line).20 The red dashed line represents a calculated spectrum based on the multiconfiguration time-dependent Hartree method and Hamiltonians as reported in ref. 23. The geometric specifications (θ = 50°, dππ = 3.4 Å) as indicated in Fig. 1 were used for the calculations. | ||
The thus enforced divergent aggregate structure of n-mer 1 directly affects the UV/vis spectrum: the most bathochromic absorption band is shifted from 536 nm (self-assembled π-stack of PBI 4)20 to 524 nm (foldamer π-stack of n-mer 1) which implies a reduced excitonic coupling ε. Moreover, a significant increase of the intensity of the most bathochromic band (and concomitantly the ratio of the absorption maxima Amax1/Amax2) for n-mer 1 compared to the self-assembled PBI aggregates is observed. Both the intensity increase of the lower energy transition band and the smaller value of the coupling ε are indeed predicted by exciton coupling theory25 for an increase of the rotational offset from φ = 30° to φ = 50°. For a quantitative analysis we have carried out MCTDH calculations for the geometry-optimized folded octamer structure shown in Fig. 1c, i.e. with eight closely π-stacked PBI dyes (d = 3.4 Å) at a rotational angle of φ = 50°. The result of this calculation is quite impressive as it shows an almost perfect fit with the measured UV/vis spectrum for n-mer 1 (and a considerable difference to the experimental UV/vis spectrum of self-assembled PBI 4a, see Fig. 6). As expected, the increased rotational offset afforded a reduced value for the coupling ε = 0.0475 eV compared to self-assembled PBIs 4a,b (0.065 eV24).
Conclusions
To conclude, a novel foldamer system has been introduced for which the folding process is driven by π–π-stacking interactions between PBI dyes that are appended to an oligophenylene ethynylene backbone. This dye-containing oligomer folds in solvents of low polarity into a helical structure driven by π–π stacking interactions of intrastrand perylene dyes. Despite the presence of an OPE backbone, this foldamer is quite different from foldamers reported by Moore and co-workers because in our system the OPE backbone served only as a semi-rigid backbone whose conformation is directed by PBI–PBI stacking interactions. Accordingly, our new foldamer features side-chain induced folding26 instead of main-chain (backbone) induced folding. With this feature, our foldamer concept is not only from the structural point of view but also from the thermodynamic one more closely related to nucleic acids whose sugar-phosphate backbone is folded by the π–π-stacking (and hydrogen-bonding) of nucleobases.Due to the presence of well-understood vibronically structured absorption and emission bands for the monomeric PBI dyes, the presented foldamer system serves as an excellent model for the elucidation of π–π interactions and the geometry of the folded state. Thus, the optical properties of the folded system were quite distinct from those of the unfolded state and those of π-stacks derived from self-assembled PBIs, revealing a unique aggregate geometry enforced by the rigid scaffold. We envision that nanoscale architectures like foldamers 1 will become important photofunctional units for a broad variety of fundamental studies, e.g. in the field of artificial photosynthesis,27 and possibly even for sensor technology and for advanced molecular materials for organic electronics and photovoltaics.
Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft within the research training school GRK 1221 is gratefully acknowledged.Notes and references
- J. D. Watson and F. H. C. Crick, Nature, 1953, 171, 737 CrossRef CAS.
- (a) G. McDermott, S. M. Prince, A. A. Freer, A. M. Hawthornthwaite-Lawless, M. Z. Papiz, R. J. Cogdell and N. W. Isaacs, Nature, 1995, 374, 517 CrossRef CAS; (b) T. Pullerits and V. Sundström, Acc. Chem. Res., 1996, 29, 381 CrossRef CAS; (c) X. Hu, T. Ritz, A. Damjanović, F. Autenrieth and K. Schulten, Q. Rev. Biophys., 2002, 35, 1 CrossRef CAS.
- (a) D. M. Bassani, J.-M. Lehn, G. Baum and D. Fenske, Angew. Chem., Int. Ed. Engl., 1997, 36, 1845 CrossRef CAS; (b) D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, Chem. Rev., 2001, 101, 3893 CrossRef CAS; (c) C. Schmuck, Angew. Chem., Int. Ed., 2003, 42, 2448 CrossRef CAS; (d) E. Yashima, K. Maeda, H. Iida, Y. Furusho and K. Nagai, Chem. Rev., 2009, 109, 6102 CrossRef CAS; (e) S. Hecht and I. Huc, Foldamers, Structures, Properties and Applications, VCH, Weinheim, 2007 Search PubMed.
- (a) C. R. Ray and J. S. Moore, Adv. Polym. Sci., 2005, 177, 91 CAS; (b) R. B. Prince, J. G. Saven, P. G. Wolynes and J. S. Moore, J. Am. Chem. Soc., 1999, 121, 3114 CrossRef CAS.
- (a) I. Huc, Eur. J. Org. Chem., 2004, 17 CrossRef CAS; (b) C. Dolain, A. Grélard, M. Laguerre, H. Jiang, V. Maurizot and I. Huc, Chem.–Eur. J., 2005, 11, 6135 CrossRef CAS; (c) G. Guichard and I. Huc, Chem. Commun., 2011, 47, 5933 RSC.
- (a) E. Yashima and K. Maeda, Macromolecules, 2008, 41, 3 CrossRef CAS; (b) A. Khan, C. Kaiser and S. Hecht, Angew. Chem., Int. Ed., 2006, 45, 1878 CrossRef CAS; (c) M. Wolffs, N. Delsuc, D. Veldman, N. Vân Anh, R. M. Williams, S. C. J. Meskers, R. A. J. Janssen, I. Huc and A. P. H. J. Schenning, J. Am. Chem. Soc., 2009, 131, 4819 CrossRef CAS.
- (a) F. Würthner, Chem. Commun., 2004, 1564 RSC; (b) S. Ghosh, X.-Q. Li, V. Stepanenko and F. Würthner, Chem.–Eur. J., 2008, 14, 11343 CrossRef CAS.
- (a) L. Schmidt-Mende, A. Fechtenkötter, K. Müllen, E. Moons, R. H. Friend and J. D. MacKenzie, Science, 2001, 293, 1119 CrossRef CAS; (b) P. Peumans, S. Uchida and S. R. Forrest, Nature, 2003, 425, 158 CrossRef CAS; (c) F. Würthner and M. Stolte, Chem. Commun., 2011, 47, 5109 RSC; (d) X. W. Zhan, A. Facchetti, S. Barlow, T. J. Marks, M. A. Ratner, M. R. Wasielewski and S. R. Marder, Adv. Mater., 2011, 23, 268 CrossRef CAS.
- (a) J. M. Giaimo, A. V. Gusev and M. R. Wasielewski, J. Am. Chem. Soc., 2002, 124, 8530 CrossRef CAS; (b) T. M. Wilson, M. J. Tauber and M. R. Wasielewski, J. Am. Chem. Soc., 2009, 131, 8952 CrossRef CAS; (c) C. Hippius, I. H. M. van Stokkum, M. Gsänger, M. M. Groeneveld, R. M. Williams and F. Würthner, J. Phys. Chem. C, 2008, 112, 2476 CrossRef CAS; (d) D. Veldman, S. M. A. Chopin, S. C. J. Meskers, M. M. Groeneveld, R. M. Williams and R. A. J. Janssen, J. Phys. Chem. A, 2008, 112, 5846 CrossRef CAS.
- (a) R. Varghese and H.-A. Wagenknecht, Chem. Commun., 2009, 2615 RSC; (b) T. M. Wilson, T. A. Zeidan, M. Hariharan, F. D. Lewis and M. R. Wasielewski, Angew. Chem., Int. Ed., 2010, 49, 1 Search PubMed.
- (a) J. Hernando, P. A. J. de Witte, E. M. H. P. van Dijk, J. Korterik, R. J. M. Nolte, A. E. Rowan, M. F. García-Parajó and N. F. van Hulst, Angew. Chem., Int. Ed., 2004, 43, 4045 CrossRef CAS; (b) P. A. J. de Witte, J. Hernando, E. E. Neuteboom, E. M. H. P. van Dijk, S. C. J. Meskers, R. A. J. Janssen, N. F. van Hulst, R. J. M. Nolte, M. F. García-Parajó and A. E. Rowan, J. Phys. Chem. B, 2006, 110, 7803 CrossRef CAS.
- For other types of π-stacked model systems, see: (a) R. Bhosale, R. S. K. Kishore, V. Ravikumar, O. Kel, E. Vauthey, N. Sakai and S. Matile, Chem. Sci., 2010, 1, 357 RSC; (b) R. Bhosale, J. Míšek, N. Sakai and S. Matile, Chem. Soc. Rev., 2010, 39, 138–149 RSC; (c) J. K. Klosterman, Y. Yamauchi and M. Fujita, Chem. Soc. Rev., 2009, 38, 1714 RSC.
- J. M. Tauber, F. R. Kelly, J. M. Giaimo, B. Rybtchinski and M. R. Wasielewski, J. Am. Chem. Soc., 2006, 128, 1782 CrossRef.
- M. Berubé and D. Poirier, Org. Lett., 2004, 6, 3127 CrossRef.
- Q. Zhou, P. J. Carroll and T. M. Swager, J. Org. Chem., 1994, 59, 1294 CrossRef CAS.
- U. H. F. Bunz, Chem. Rev., 2000, 100, 1605 CrossRef CAS.
- (a) S. H. Gross, Mass Spectrometry, Springer-Verlag, Berlin, Heidelberg, 2004 Search PubMed; (b) R. J. Goldschmitt, S. J. Wetzel, S. W. R. Blair and C. M. Guttman, J. Am. Soc. Mass Spectrom., 2000, 11, 1095 CrossRef.
- H.-G. Elias, Polymere: Von Monomeren und Makromolekülen zu Werkstoffen, Hüthig & Wepf Verlag: Zug, Heidelberg, Oxford, 1996 Search PubMed.
- (a) R. Arakawa, S. Watanabe and T. Fukuo, Rapid Commun. Mass Spectrom., 1999, 13, 1059 CrossRef CAS; (b) M. F. E. Nielen and S. Malucha, Rapid Commun. Mass Spectrom., 1997, 11, 1194 CrossRef CAS.
- (a) V. Dehm, Z. Chen, U. Baumeister, P. Prins, L. D. A. Siebbeles and F. Würthner, Org. Lett., 2007, 9, 1085 CrossRef CAS; (b) Z. Chen, V. Stepanenko, V. Dehm, P. Prins, L. D. A. Siebbeles, J. Seibt, P. Marquetand, V. Engel and F. Würthner, Chem.–Eur. J., 2007, 13, 436 CrossRef CAS.
- J. J. Han, A. D. Shaller, W. Wang and A. D. Q. Li, J. Am. Chem. Soc., 2008, 130, 6974 CrossRef CAS.
- C. N. Pace, B. A. Shirley and J. A. Thomson, Protein Structure: A Practical Approach, IRL Press, New York, 1989 Search PubMed.
- J. Seibt, T. Winkler, K. Renziehausen, V. Dehm, F. Würthner, H.-D. Meyer and V. Engel, J. Phys. Chem. A, 2009, 113, 13475 CrossRef CAS.
- R. Fink, J. Seibt, V. Engel, M. Renz, M. Kaupp, S. Lochbrunner, H.-M. Zhao, J. Pfister, F. Würthner and B. Engels, J. Am. Chem. Soc., 2008, 130, 12858 CrossRef CAS.
- M. Kasha, H. R. Rawls and M. A. El-Bayoumi, Pure Appl. Chem., 1965, 11, 371 CrossRef CAS.
- (a) F. A. Syud, H. E. Stanger and S. H. Gellman, J. Am. Chem. Soc., 2001, 123, 8667 CrossRef CAS; (b) X. Zhao, M.-X. Jia, X.-K. Jiang, L.-Z. Wu, Z.-T. Li and G.-J. Chen, J. Org. Chem., 2004, 69, 270 CrossRef CAS; (c) D. Haldar, H. Jiang, J.-M. Léger and I. Huc, Angew. Chem., Int. Ed., 2006, 45, 5483 CrossRef CAS; (d) E. B. Berda, E. J. Foster and E. W. Meijer, Macromolecules, 2010, 43, 1430 CrossRef CAS.
- M. R. Wasielewski, Acc. Chem. Res., 2009, 42, 1910 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details, mass and DOSY NMR data, additional UV/vis studies, and Keq and ΔG values obtained for folding of 1. See DOI: 10.1039/c1sc00435b |
| This journal is © The Royal Society of Chemistry 2011 |