Chromium(V)-oxo and chromium(III)-superoxo complexes bearing a macrocyclic TMC ligand in hydrogen atom abstraction reactions†
Jaeheung
Cho‡
a,
Jaeyoung
Woo‡
a,
Jung
Eun Han
a,
Minoru
Kubo
b,
Takashi
Ogura
b and
Wonwoo
Nam
*a
aDepartment of Bioinspired Science, Department of Chemistry and Nano Science, Ewha Womans University, Seoul 120–750, Korea. E-mail: wwnam@ewha.ac.kr; Fax: (+82) 2-3277-4441; Tel: (+82) 2-3277-2392
bPicobiology Institute, Graduate School of Life Science, University of Hyogo, Koto 3-2-1, Kamigori-cho, Ako-gun, Hyogo 678-1297, Japan
First published on 4th August 2011
Abstract
A Cr(V)-oxo complex bearing a macrocyclic TMC ligand, [CrV(TMC)(O)(OCH3)]2+, was synthesized, isolated, and characterized by various physicochemical methods, including UV-vis, ESI-MS, resonance Raman, EPR and X-ray analysis. The reactivity of the Cr(V)-oxo complex was investigated in C–H and O–H bond activation reactions. The reactivity of a Cr(III)-superoxo complex, [CrIII(TMC)(O2)(Cl)]+, was investigated in O–H bond activation reactions as well. By comparing reactivities of the Cr(III)-superoxo and Cr(V)-oxo complexes under the identical reaction conditions, we were able to demonstrate that the Cr(III)-superoxo complex is more reactive than the Cr(V)-oxo complex in the activation of C–H and O–H bonds. The present results provide strong evidence that under certain circumstances, metal-superoxo species can be an alternative oxidant for high-valent metal-oxo complexes in oxygenation reactions.
Introduction
Dioxygen is essential in life processes, and metalloenzymes activate dioxygen to carry out a variety of biological reactions. Oxygen-coordinating metal intermediates, such as metal-superoxo, -peroxo, -hydroperoxo, and -oxo species, have been invoked as reactive species in the catalytic oxygenation reactions.1 Among the metal-oxygen intermediates, the chemistry of high-valent metal-oxo species has been intensively studied using biomimetic compounds over the past several decades. For example, high-valent iron(IV)-oxo complexes of heme and nonheme ligands have been synthesized as chemical models of cytochrome P450 and nonheme iron enzymes, respectively, and have shown reactivities in various oxidation reactions, including alkane hydroxylation and olefin epoxidation.2,3Metal-superoxo intermediates have also attracted much attention recently in the communities of bioinorganic and biological chemistry, since the intermediates have been invoked as reactive species in the C–H bond activation of substrates by nonheme iron and copper enzymes.4,5 In biomimetic studies, synthetic Cu(II)-superoxo complexes have shown reactivities in ligand oxidation and weak O–H and N–H bond activation reactions.6 Very recently, we have demonstrated that a chromium(III)-superoxo complex bearing a macrocyclic ligand, [CrIII(TMC)(O2)(Cl)]+ (1) (TMC = 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane), is capable of activating weak C–H bonds of hydrocarbonsvia a hydrogen atom (H-atom) abstraction mechanism (Scheme 1).7,8 More recently, an iron(III)-superoxo intermediate, [FeIII(TMC)(O2)]2+, has been proposed as an active oxidant that abstracts an H-atom from weak C–H bonds of substrates, resulting in the generation of its corresponding iron(IV)-oxo complex, [FeIV(TMC)(O)]2+.9 These results suggest that in addition to the high-valent metal-oxo species, metal-superoxo complexes are potent oxidants capable of activating C–H (or O–H) bonds of substrates to give oxygenated products. However, to the best of our knowledge, reactivities of the high-valent metal-oxo and metal-superoxo complexes have never been compared directly in those reactions.10 We therefore decided to synthesize a Cr(V)-oxo complex bearing a TMC ligand and compare its reactivity with the previously reported Cr(III)-superoxo complex, 1, in oxidation reactions (Scheme 1). Herein, we report the synthesis, isolation, and spectroscopic and structural characterization of [CrV(TMC)(O)(OCH3)]2+ (2) and a reactivity comparison of the Cr(V)-oxo and Cr(III)-superoxo complexes in C–H and O–H bond activation reactions.
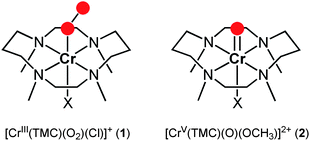 | ||
| Scheme 1 A schematic drawing of the structures of 1 and 2. | ||
Results and discussion
We first synthesized a chromium(V)-oxo complex bearing a TMC ligand, [CrV(TMC)(O)(OCH3)]2+ (2).11 The reaction of [Cr(TMC)(CH3CN)]2+ with 2 equiv. of iodosylbenzene (PhIO), dissolved in CH3OH, in CH3CN at −10 °C produces a dark orange intermediate 2 (see ESI†, Experimental Section and Fig. S1 for the synthesis and a structure of the starting [Cr(TMC)(CH3CN)]2+ complex, respectively). The intermediate persisted for several days at −10 °C, and the greater thermal stability of 2 allowed us to isolate crystals and use the isolated crystals in spectroscopic and structural characterization and reactivity studies.The UV-vis spectrum of 2 shows intense absorption bands at 353 (ε = 4800 M−1 cm−1) and 446 nm (ε = 2000 M−1 cm−1) and a shoulder at ∼520 nm (ε = 550 M−1 cm−1) (Fig. 1a). The electrospray ionization mass spectrum (ESI-MS) of 2 exhibits a prominent signal at a mass-to-charge (m/z) ratio of 177.6 (Fig. 1b), whose mass and isotope distribution pattern correspond to [Cr(TMC)(O)(OCH3)]2+ (calculated m/z 177.6) (Fig. 1b, inset). When 2 was prepared with isotopically labelled PhI18O in the presence of H218O, the mass peak corresponding to 2 shifted to m/z 178.6 (Fig. 1b, inset), indicating that 2 contains an oxygen atom in it. The X-band electron paramagnetic resonance (EPR) spectrum of a frozen CH3CN solution of 2 exhibits a strong signal at g = 1.98 with 41 G maximum-to-minimum separation (Fig. 1a, inset), which is typical for Cr(V) (S = 1/2).11 The resonance Raman spectrum of 2, measured in CD3CN at −20 °C with 407 nm laser excitation, exhibits an isotope-sensitive band at 907 cm−1, which shifted to 872 cm−1 when 2-18O was generated with isotopically labelled PhI18O in the presence of H218O in CD3CN (Fig. 1c). The observed isotopic shift of −35 cm−1 with 18O-substitution is in agreement with the calculated value (Δνcalc = −39 cm−1) for the Cr–O diatomic harmonic oscillator. The observed Cr–O frequency at 907 cm−1 is lower than those reported in Cr-oxo complexes bearing salen (∼1000 cm−1, infrared),11bporphyrin (∼1025 cm−1, infrared),12aphthalocyanine (∼1041 cm−1, infrared),12b and corrole (∼1025 cm−1, resonance Raman).11i
![(a) UV-vis spectrum of [CrV(TMC)(O)(OCH3)]2+ (2) in CH3CN at 25 °C. Inset shows the X-band EPR spectrum of 2 (g = 1.98) in frozen CH3CN at 4.3 K. Instrumental parameters: microwave power = 1.007 mW, frequency = 9.646 GHz, sweep width = 0.4 T, modulation amplitude = 1 mT. (b) ESI-MS of 2 in CH3CN at 25 °C. Insets show the observed isotope distribution patterns for 2-16O (lower) and 2-18O (upper). (c) Resonance Raman spectra of 2 (8 mM) obtained upon excitation at 407 nm at −20 °C; 2-16O (red line) and 2-18O (blue line) were recorded in CD3CN. The peaks marked with “s” are ascribed to solvent bands.](/image/article/2011/SC/c1sc00386k/c1sc00386k-f1.gif) | ||
| Fig. 1 (a) UV-vis spectrum of [CrV(TMC)(O)(OCH3)]2+ (2) in CH3CN at 25 °C. Inset shows the X-band EPR spectrum of 2 (g = 1.98) in frozen CH3CN at 4.3 K. Instrumental parameters: microwave power = 1.007 mW, frequency = 9.646 GHz, sweep width = 0.4 T, modulation amplitude = 1 mT. (b) ESI-MS of 2 in CH3CN at 25 °C. Insets show the observed isotope distribution patterns for 2-16O (lower) and 2-18O (upper). (c) Resonance Raman spectra of 2 (8 mM) obtained upon excitation at 407 nm at −20 °C; 2-16O (red line) and 2-18O (blue line) were recorded in CD3CN. The peaks marked with “s” are ascribed to solvent bands. | ||
Single crystals of 2 were grown from CH3CN/diethyl ether at −30 °C, and a crystallographic analysis revealed that 2-(ClO4)2 is a mononuclear chromium-oxo complex in a distorted octahedral geometry (Fig. 2; see ESI†, Tables S1 and S2). Notably, the Cr–O bond length (1.604(3) Å) of 2 is longer than those of other Cr(IV or V)-oxo complexes (1.49–1.57 Å),11a,11b,11h,12a,13 which is in line with the low Cr–O stretching vibration observed in the resonance Raman spectrum of 2 (vide supra). As illustrated by the space-filling representation in Fig. 2b, the oxo ligand is nestled in a bed of H atoms from C–H bonds of the TMC ligand, as observed in [FeIV(TMC)(O)(CH3CN)]2+.14 Further, as suggested in [FeIV(TMC)(O)(CH3CN)]2+,14 the thermal stability of 2 is due to the steric encumbrance for the access of external substrates to the chromium-oxo moiety. All four N-methyl groups of the TMC ligand in 2 point away from the oxo group (Fig. 2), as observed in the [FeIV(TMC)(O)(CH3CN)]2+ structure.14 Thus, the structure of 2 resembles that of the iron(IV)-oxo analogue. Another interesting structural phenomenon is a relatively short Cr–O2 bond length (1.765(3) Å) and the linearity of the Cr–O2–C9 bond angle (176.7(3)°). Although metal (M)–O–Me groups are usually bent, linear M–O–Me angles with short M–O bond length have been observed in several metal-alkoxide bonds with multiple-bond character, where an oxygen atom acts as both σ- and π-donors.15 Consequently, the relatively long Cr–O bond of 2, compared to other Cr-oxo complexes, results from the strongly binding methoxide ligand trans to the Cr-oxo group.
![(a) ORTEP plot of [CrV(TMC)(O)(OCH3)]2+ (2) with 30% probability thermal ellipsoid. Hydrogen atoms are omitted for clarity. (b) Space-filling representation of 2, derived from the single crystal structure determination. Selected bond lengths (Å) and angles (°): Cr–O1 1.604(3), Cr–O2 1.765(3), Cr–N1 2.122(2), Cr–N2 2.148(2); O1–Cr–O2 178.39(13), Cr–O2–C9 176.7(3).](/image/article/2011/SC/c1sc00386k/c1sc00386k-f2.gif) | ||
| Fig. 2 (a) ORTEP plot of [CrV(TMC)(O)(OCH3)]2+ (2) with 30% probability thermal ellipsoid. Hydrogen atoms are omitted for clarity. (b) Space-filling representation of 2, derived from the single crystal structure determination. Selected bond lengths (Å) and angles (°): Cr–O1 1.604(3), Cr–O2 1.765(3), Cr–N1 2.122(2), Cr–N2 2.148(2); O1–Cr–O2 178.39(13), Cr–O2–C9 176.7(3). | ||
We then compared the reactivities of the chromium(V)-oxo and chromium(III)-superoxo complexes in the activation of C–H and O–H bonds. First, the reactivity of the Cr(III)-superoxo complex, 1, was investigated in O–H bond activation reactions; the reactivity of 1 in C–H bond activation reactions was reported previously.7 Upon addition of 2,6-di-tert-butylphenol (DTBP) to 1 in CH3CN at −40 °C, 1 was converted to the chromium(III)-hydroxo complex, [CrIII(TMC)(OH)(Cl)]+,7 and showed pseudo-first-order decay as monitored by a UV-vis spectrophotometer (see ESI†, Fig. S2a). Pseudo-first-order fitting of the kinetic data yielded the kobs value of 9.1 × 10−3s−1. The rate constant increased proportionally with the substrate concentration, affording a second-order rate constant, k2, of 4.7 × 10−1 M−1s−1 at −40 °C (see ESI†, Fig. S2b). Product analysis of the resulting solution revealed that 2,6-di-tert-butyl-1,4-benzoquinone (DTBQ) (45 ± 10%) was produced in the oxidation of DTBP, as reported in the oxidation of DTBP by other metal-superoxo complexes.16
Further mechanistic studies with para-substituted 2,6-di-tert-butylphenols (para-Y-2,6-tBu2-C6H2OH; Y = OMe, Me, H, Br, CN) revealed that the second-order rate constants were dependent markedly on the electron-donating and -withdrawing properties of the para-substitutents (Table 1).17 By plotting the rates as a function of σP+ of the para-substituents, a good linear correlation was obtained with a negative Hammett ρ value of −3.8 (Fig. 3a). Further, we observed a good linear correlation when the rates were plotted against the phenol O–H bond dissociation energies (BDE) (Fig. 3b) and the phenol O–H redox potentials (see ESI†, Fig. S3). Such a linear relationship in the Hammett plot and the dependence of the rate constants on the O–H BDE of substrates implicates an H-atom abstraction as the rate-determining step in the oxidation of phenol O–H bonds by 1.18
| Y | σp+ | BDE (kcal mol−1)a | k 2 (M−1s−1) | log k2 |
|---|---|---|---|---|
| a Ref. 17. b Due to the low reactivity of para-CN-2,6-tBu2-C6H2OH, the k2 value was determined at −10 °C and normalized using the Eyring equation. The determined second-order rate constant, k2, at −10 °C was 8.0 × 10−2 M−1s−1. | ||||
| OMe | –0.78 | 78.31 | 1.3 × 103 | 3.11 |
| Me | –0.31 | 81.02 | 1.6 × 10 | 1.20 |
| H | 0 | 82.8 | 4.7 × 10−1 | –0.33 |
| Br | 0.15 | - | 2.7 × 10−1 | –0.57 |
| CNb | 0.66 | 84.24 | 4.2 × 10−3 | –2.38 |
![(a) Hammett plot for the oxidation of para-substituted 2,6-di-tert-butylphenols, p-Y-2,6-tBu2-C6H2OH (Y = OMe, Me, H, Br, CN), by [CrIII(TMC)(O2)(Cl)]+ (1) (2 mM) in CH3CN at −40 °C. (b) Plot of log k2 of 1 against O–H bond dissociation energy (BDE) of para-Y-2,6-tBu2-C6H2OH in CH3CN at −40 °C.](/image/article/2011/SC/c1sc00386k/c1sc00386k-f3.gif) | ||
| Fig. 3 (a) Hammett plot for the oxidation of para-substituted 2,6-di-tert-butylphenols, p-Y-2,6-tBu2-C6H2OH (Y = OMe, Me, H, Br, CN), by [CrIII(TMC)(O2)(Cl)]+ (1) (2 mM) in CH3CN at −40 °C. (b) Plot of log k2 of 1 against O–H bond dissociation energy (BDE) of para-Y-2,6-tBu2-C6H2OH in CH3CN at −40 °C. | ||
We then investigated the reactivity of 2 in C–H and O–H bond activation with substrates such as cyclohexadiene (CHD) and DTBP. Upon addition of the substrates to the solution of 2 in CH3CN at −10 °C for CHD and at −40 °C for DTBP, the intermediate remained intact under these conditions. We therefore carried out the reactions at a high temperature (i.e., 30 °C for C–H bond activation and 0 °C for O–H bond activation). Upon addition of CHD to 2 in CH3CN at 30 °C, the characteristic UV-vis absorption bands of 2 disappeared with a first-order decay profile (Fig. 4a), and the pseudo-first-order rate constants increased proportionally with the concentration of CHD (k2 = 1.0 × 10−3 M−1s−1 at 30 °C) (Fig. 4b). Product analysis of the resulting solution revealed the formation of benzene (90 ± 10%) as a product, as observed in the oxidation of CHD by metal-oxo and metal-superoxo complexes.7,18,19 In addition, [CrIII(TMC)(OH)(CH3CN)]2+ was found in the reaction solutions as a decomposed product of 2 (see ESI†, Fig. S4 for ESI-MS analysis). By comparing the second-order rate constants of 1 (k2 = 1.7 × 10−1 M−1s−1 at −10 °C)7 and 2 (k2 = 1.0 × 10−3 M−1s−1 at 30 °C) in the CHD oxidation reaction, the reactivity of the Cr(III)-superoxo species (1) is much greater than that of the Cr(V)-oxo species (2) in C–H bond activation reactions. In the O–H bond activation reactions, rate constants in the reactions of 2 and DTBP were not able to be determined due to the low reactivity. Alternatively, we were able to follow the kinetics of the reaction of 2 with para-MeO-2,6-di-tert-butylphenol (p-MeO-DTBP). Upon addition of p-MeO-DTBP to 2 in CH3CN at 0 °C, the pseudo-first-order rate constants increased proportionally with the concentration of p-MeO-DTBP (k2 = 2.3 × 10−2 M−1s−1) (Fig. 5). Product analysis of the resulting solution revealed that DTBQ (90 ± 10%) was produced as a product.16 Then, [CrIII(TMC)(OH)(CH3CN)]2+ was found in the reaction solutions as a decomposed product of 2. By comparing the kinetic data of 1 (k2 = 1.3 × 103 M−1s−1 at −40 °C) and 2 (k2 = 2.3 × 10−2 M−1s−1 at 0 °C) in the p-MeO-DTBP oxidation reaction (Table 1 and Fig. 5), the reactivity of the Cr(III)-superoxo species (1) is much greater than that of the Cr(V)-oxo species (2) in O–H bond activation reactions. Although the present results demonstrate clearly that 1 is a much stronger oxidant than 2, we should also mention that the low reactivity of 2 might result from the steric encumbrance for the access of external substrates to the chromium-oxo moiety (vide supra). That is, the access of substrates toward the oxo group of 2 is much more difficult than for the case of the superoxo group of 1, as shown in the space-filling models of 1 and 2 (see ESI†, Fig. S5). Together with the steric effect, a strong binding of methoxide to Cr(V) may decrease the electrophilicity of the Cr(V)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety in the C–H and O–H bond activation reactions, which is an axial ligand effect as reported in the C–H bond activation and oxo-transfer reactions by heme and nonheme metal-oxo complexes.20
O moiety in the C–H and O–H bond activation reactions, which is an axial ligand effect as reported in the C–H bond activation and oxo-transfer reactions by heme and nonheme metal-oxo complexes.20
![Reaction of [CrV(TMC)(O)(OCH3)]2+ (2) with 1,4-cyclohexadiene (CHD) in CH3CN at 30 °C. (a) UV-vis spectral changes of 2 (2 mM) upon addition of 400 equiv. of CHD (0.2 cm path quartz cell). Inset shows the time course of the absorbance at 446 nm. (b) Plot of kobs against CHD concentration to determine a second-order rate constant.](/image/article/2011/SC/c1sc00386k/c1sc00386k-f4.gif) | ||
| Fig. 4 Reaction of [CrV(TMC)(O)(OCH3)]2+ (2) with 1,4-cyclohexadiene (CHD) in CH3CN at 30 °C. (a) UV-vis spectral changes of 2 (2 mM) upon addition of 400 equiv. of CHD (0.2 cm path quartz cell). Inset shows the time course of the absorbance at 446 nm. (b) Plot of kobs against CHD concentration to determine a second-order rate constant. | ||
![Kinetic studies of the reactions of [CrIII(TMC)(O2)(Cl)]+ (1) and [CrV(TMC)(O)(OCH3)]2+ (2) with para-MeO-2,6-tBu2-C6H2OH (p-MeO-DTBP). Plots of kobs against the concentration of p-MeO-DTBP to determine second-order rate constants for the reactions of 1 at −40 °C (red line with ▲) and 2 at 0 °C (blue line with ●) in CH3CN.](/image/article/2011/SC/c1sc00386k/c1sc00386k-f5.gif) | ||
| Fig. 5 Kinetic studies of the reactions of [CrIII(TMC)(O2)(Cl)]+ (1) and [CrV(TMC)(O)(OCH3)]2+ (2) with para-MeO-2,6-tBu2-C6H2OH (p-MeO-DTBP). Plots of kobs against the concentration of p-MeO-DTBP to determine second-order rate constants for the reactions of 1 at −40 °C (red line with ▲) and 2 at 0 °C (blue line with ●) in CH3CN. | ||
Conclusions
We have shown the synthesis, isolation, and spectroscopic and structural characterization of a new Cr(V)-oxo complex bearing a macrocyclic TMC ligand. The reactivity of the intermediate was investigated in C–H and O–H bond activation reactions. By comparing the reactivities of the Cr(III)-superoxo and Cr(V)-oxo complexes under the identical reaction conditions, we were able to demonstrate that the Cr(III)-superoxo complex is more reactive than the Cr(V)-oxo analogue in the activation of C–H and O–H bonds. The present work, therefore, provides the first direct reactivity comparison of metal-superoxo and high-valent metal-oxo species that have been frequently invoked as key intermediates in the catalytic cycles of nonheme iron and copper enzymes. Finally, the present results lead us to propose that under certain circumstances, metal-superoxo species can be viable and alternative oxidants for high-valent metal-oxo complexes in oxygenation reactions.Experimental section
Synthesis of [Cr(TMC)(CH3CN)](ClO4)2
[Cr(TMC)(Cl)](Cl)·2CH3CN (0.092 g, 0.2 mmol) was dissolved in CH3CN (3 mL), to which was added NaClO4 (0.098 g, 0.8 mmol). The reaction solution was allowed to stand for a few days under N2. After slow diffusion of diethyl ether to the resulting solution, blue crystals suitable for X-ray analysis were isolated (see ESI†, Fig. S1). Crystalline yield: 0.067 g (61%). UV-vis (CH3CN): λmax (ε) = 659 nm (80 M−1 cm−1).Synthesis of [Cr(TMC)(O)(OCH3)](ClO4)2 (2-(ClO4)2)
Treatment of [Cr(TMC)(CH3CN)](ClO4)2 (8.8 mg, 0.016 mmol) in CH3CN (1 mL) with 2 equiv. PhIO in CH3OH (200 μL) at −30 °C afforded the formation of a dark orange solution. Slow diffusion of diethyl ether to the resulting solution afforded dark orange crystals suitable for X-ray analysis. Crystalline yield: 0.0059 g (56%). Spectroscopic data, including UV-vis, EPR, ESI-MS, and resonance Raman, were reported in Fig. 1. [Cr(TMC)(18O)(OCH3)]2+ (2-18O) was prepared by adding 2 equiv. PhIO, dissolved in CH3OH (200 μL) in the presence of H218O (10 μL), to a solution containing [Cr(TMC)(CH3CN)](ClO4)2 (8.8 mg, 0.016 mmol) in CH3CN (1 mL) at −30 °C.Reactivity studies
All reactions were run monitoring UV-vis spectral changes of reaction solutions, and rate constants were determined by fitting the changes in absorbance at 549 nm for 1 and 446 nm for 2. Reactions were run at least in triplicate, and the data reported represent the average of these reactions. Complex 1 was prepared by reacting [Cr(TMC)(Cl)]+ with excess of O2 at −40 °C and used directly in reactivity studies, such as the oxidation of para-substituted 2,6-di-tert-butylphenols (para-Y-2,6-tBu2-C6H2OH; Y = OMe, Me, H, Br, CN) under stoichiometric conditions in CH3CN at the given temperature. The isolated crystalline sample of 2 was used in kinetic studies, such as the oxidation of CHD under stoichiometric conditions in CH3CN at 30 °C. For the stopped-flow experiments, all reaction traces were collected at 549 nm, using a 1 cm optical path length at −40 °C. The raw kinetic data were treated with KinetAsyst 3 (Hi-Tech Scientific) and Specfit/32 Global Analysis System software from Spectrum Software Associates. The purity of substrates was checked with GC and GC-MS prior to use. Products were analyzed by injecting the reaction mixture directly into GC and GC-MS. Products were identified by comparing with authentic samples, and product yields were determined by comparison against standard curves prepared with authentic samples and using decane as an internal standard.X-ray crystallography
Crystal data for 2-(ClO4)2: C15H35Cl2CrN4O10, orthorhombic, Pnma, Z = 4, a = 14.2150(4) Å, b = 9.7664(3) Å, c = 16.4588(5) Å, V = 2284.96(12) Å3, μ = 0.794 mm−1, ρc = 1.611 g cm−3, R1 = 0.0486, wR2 = 0.2095 for 2378 unique reflections, 166 variables. CCDC reference number 812941.Acknowledgements
WN at EWU acknowledges the financial support from the NRF/MEST of Korea through the CRI, GRL (2010-00353), and WCU (R31-2008-000-10010-0) and the 2011 KRICT OASIS Project. JC at EWU thanks the National Research Foundation of Korea (NRF-2010-1054-1-2). The research at UH was supported by Grant-in-Aid for scientific research on priority area (No. 477) from MEXT, Japan to TO (22018026).Notes and references
- W. Nam, Acc. Chem. Res., 2007, 40, 465 CrossRef CAS , and review articles in the special issue.
- (a) P. R. Ortiz de Montellano, Cytochrome P450: Structure, Mechanism, and Biochemistry, 3rd edn, Kluwer Academic/Plenum Publishers, New York, 2005 Search PubMed; (b) I. G. Denisov, T. M. Makris, S. G. Sligar and I. Schlichting, Chem. Rev., 2005, 105, 2253–2277 CrossRef CAS; (c) R. van Eldik, Coord. Chem. Rev., 2007, 251, 1649–1662 CrossRef CAS; (d) S. Shaik, H. Hirao and D. Kumar, Acc. Chem. Res., 2007, 40, 532–542 CrossRef CAS.
- (a) P. C. A. Bruijnincx, G. van Koten and R. J. M. Klein Gebbink, Chem. Soc. Rev., 2008, 37, 2716–2744 RSC; (b) C. Krebs, D. G. Fujimori, C. T. Walsh and J. M. Bollinger, Jr, Acc. Chem. Res., 2007, 40, 484–492 CrossRef CAS; (c) W. Nam, Acc. Chem. Res., 2007, 40, 522–531 CrossRef CAS; (d) M. M. Abu-Omar, A. Loaiza and N. Hontzeas, Chem. Rev., 2005, 105, 2227–2252 CrossRef CAS.
- (a) W. A. van der Donk, C. Krebs and J. M. Bollinger, Jr, Curr. Opin. Struct. Biol., 2010, 20, 673–683 Search PubMed; (b) A. Mukherjee, M. A. Cranswick, M. Chakrabarti, T. K. Paine, K. Fujisawa, E. Münck and L. Que, Jr, Inorg. Chem., 2010, 49, 3618–3628 Search PubMed; (c) J. M. Bollinger, Jr and C. Krebs, Curr. Opin. Chem. Biol., 2007, 11, 151–158 CrossRef.
- (a) S. T. Prigge, B. A. Eipper, R. E. Mains and L. M. Amzel, Science, 2004, 304, 864–867 CrossRef CAS; (b) P. Chen and E. I. Solomon, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 13105–13110 CrossRef CAS; (c) J. P. Klinman, J. Biol. Chem., 2006, 281, 3013–3016 CAS; (d) M. Rolff and F. Tuczek, Angew. Chem., Int. Ed., 2008, 47, 2344–2347 CrossRef CAS.
- (a) D. Maiti, H. C. Fry, J. S. Woertink, M. A. Vance, E. I. Solomon and K. D. Karlin, J. Am. Chem. Soc., 2007, 129, 264–265 CrossRef CAS; (b) T. Fujii, S. Yamaguchi, S. Hirota and H. Masuda, Dalton Trans., 2008, 164–170 RSC; (c) A. Kunishita, M. Kubo, H. Sugimoto, T. Ogura, K. Sato, T. Takui and S. Itoh, J. Am. Chem. Soc., 2009, 131, 2788–2789 CrossRef CAS; (d) R. L. Peterson, R. A. Himes, H. Kotani, T. Suenobu, L. Tian, M. A. Siegler, E. I. Solomon, S. Fukuzumi and K. D. Karlin, J. Am. Chem. Soc., 2011, 133, 1702–1705 Search PubMed.
- J. Cho, J. Woo and W. Nam, J. Am. Chem. Soc., 2010, 132, 5958–5959 CrossRef CAS.
- Bakac and co-workers reported reactivities of Cr(III)-superoxo species in H-atom transfer reactions using transition-metal hydrides and hydroperoxides as substrates in aqueous solution: (a) A. Bakac, Coord. Chem. Rev., 2006, 250, 2046–2058 CrossRef CAS; (b) A. Bakac, J. Am. Chem. Soc., 1997, 119, 10726–10731 CrossRef CAS.
- Y.-M. Lee, S. Hong, Y. Morimoto, W. Shin, S. Fukuzumi and W. Nam, J. Am. Chem. Soc., 2010, 132, 10668–10670 CrossRef CAS.
- The reactivity of CrIVaqO2+ is reported to be 102-fold greater than that of CrIIIaqOO2+ in the oxidation of L(H2O)RhOOH in aqueous solution. Theopold and co-workers also reported crystal structures of CrIV-oxo and CrIII-superoxo complexes bearing a hydrotrispyrazolylborate ligand and demonstrated that the former is reactive enough to activate C–H bonds of cyclohexadiene and 9,10-dihydroanthracene, whereas the latter is stable at room temperature: (a) M. J. Vasbinder and A. Bakac, Inorg. Chem., 2007, 46, 2921–2928 Search PubMed; (b) K. Qin, C. D. Incarvito, A. L. Rheingold and K. H. Theopold, Angew. Chem., Int. Ed., 2002, 41, 2333–2335 CrossRef CAS; (c) K. Qin, C. D. Incarvito, A. L. Rheingold and K. H. Theopold, J. Am. Chem. Soc., 2002, 124, 14008–14009 CrossRef CAS.
- The synthesis and structural and spectroscopic characterization of Cr(V)-oxo complexes with salen, porphyrin, and corrole ligands have been reported previously. In addition, synthetic Cr(V)-oxo complexes have shown reactivities in oxidation reactions, including the epoxidation of olefins, and it has been proposed that Cr(V)-oxo species are reactive intermediates in the catalytic oxidation of organic substrates: (a) T. L. Siddall, N. Miyaura, J. C. Huffman and J. K. Kochi, J. Chem. Soc., Chem. Commun., 1983, 1185–1186 RSC; (b) K. Srinivasan and J. K. Kochi, Inorg. Chem., 1985, 24, 4671–4679 CrossRef CAS; (c) E. G. Samsel, K. Srinivasan and J. K. Kochi, J. Am. Chem. Soc., 1985, 107, 7606–7617 CrossRef CAS; (d) J. M. Garrison and T. C. Bruice, J. Am. Chem. Soc., 1989, 111, 191–198 CrossRef CAS; (e) J. M. Garrison, D. Ostovic and T. C. Bruice, J. Am. Chem. Soc., 1989, 111, 4960–4966 CrossRef CAS; (f) A. Bakac and W.-D. Wang, J. Am. Chem. Soc., 1996, 118, 10325–10326 CrossRef CAS; (g) H. Fujii, T. Yoshimura and H. Kamada, Inorg. Chem., 1997, 36, 1122–1127 CrossRef; (h) A. E. Meier-Callahan, H. B. Gray and Z. Gross, Inorg. Chem., 2000, 39, 3605–3607 CrossRef CAS; (i) R. S. Czernuszewicz, V. Mody, A. Czader, M. Gałęzowski and D. T. Gryko, J. Am. Chem. Soc., 2009, 131, 14214–14215 Search PubMed.
- (a) J. T. Groves, W. J. Kruper, Jr, R. C. Haushalter and W. M. Butler, Inorg. Chem., 1982, 21, 1363–1368 CrossRef CAS; (b) K. H. Nill, F. Wasgestian and A. Pfeil, Inorg. Chem., 1979, 18, 564–567 CrossRef CAS.
- (a) M. Krumpolc, B. G. DeBoer and J. Roček, J. Am. Chem. Soc., 1978, 100, 145–153 CrossRef CAS; (b) R. J. Judd, T. W. Hambley and P. A. Lay, J. Chem. Soc., Dalton Trans., 1989, 2205–2210 RSC.
- J.-U. Rohde, J.-H. In, M. H. Lim, W. W. Brennessel, M. R. Bukowski, A. Stubna, E. Münck, W. Nam and L. Que, Jr, Science, 2003, 299, 1037–1039 CrossRef CAS.
- (a) F. A. Cotton, G. Wilkinson, C. A. Murillo and M. Bochmann, Advanced Inorganic Chemistry, 6th ed., Wiley, New York, 1999, p 472 Search PubMed; (b) R. Karia, G. R. Willey and M. G. B. Drew, J. Chem. Soc., Dalton Trans., 1986, 2493–2495 RSC; (c) R. T. Jonas and T. D. P. Stack, J. Am. Chem. Soc., 1997, 119, 8566–8567 CrossRef CAS.
- (a) A. Company, S. Yao, K. Ray and M. Driess, Chem.–Eur. J., 2010, 16, 9669–9675 Search PubMed; (b) D. Maiti, D.-H. Lee, K. Gaoutchenova, C. Würtele, M. C. Holthausen, A. A. Narducci Sarjeant, J. Sundermeyer, S. Schindler and K. D. Karlin, Angew. Chem., Int. Ed., 2008, 47, 82–85 CrossRef CAS; (c) G. T. Musie, M. Wei, B. Subramaniam and D. H. Busch, Inorg. Chem., 2001, 40, 3336–3341 CrossRef CAS; (d) Y. Deng and D. H. Busch, Inorg. Chem., 1995, 34, 6380–6386 CrossRef CAS.
- G. Brigati, M. Lucarini, V. Mugnaini and G. F. Pedulli, J. Org. Chem., 2002, 67, 4828–4832 CrossRef CAS.
- (a) D. T. Y. Yiu, M. F. W. Lee, W. W. Y. Lam and T.-C. Lau, Inorg. Chem., 2003, 42, 1225–1232 CrossRef CAS; (b) D. E. Lansky and D. P. Goldberg, Inorg. Chem., 2006, 45, 5119–5125 CrossRef CAS; (c) M. S. Seo, N. H. Kim, K.-B. Cho, J. E. So, S. K. Park, M. Clémancey, R. Garcia-Serres, J.-M. Latour, S. Shaik and W. Nam, Chem. Sci., 2011, 2, 1039–1045 RSC.
- A H-atom abstraction of phenols by 1 generates [CrIII(TMC)(OOH)(Cl)]+, followed by the hydroperoxo O–O bond cleavage that leads to the formation of Cr(IV or V)-oxo species. The resulting Cr-oxo species is involved in the H-atom abstraction reaction, giving the [CrIII(TMC)(OH)(Cl)]+ product.
- (a) C. V. Sastri, J. Lee, K. Oh, Y. J. Lee, J. Lee, T. A. Jackson, K. Ray, H. Hirao, W. Shin, J. A. Halfen, J. Kim, L. Que, Jr, S. Shaik and W. Nam, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 19181–19186 CrossRef; (b) H. Hirao, L. Que, Jr, W. Nam and S. Shaik, Chem.–Eur. J., 2008, 14, 1740–1756 CrossRef CAS; (c) S. N. Dhuri, M. S. Seo, Y.-M. Lee, H. Hirao, Y. Wang, W. Nam and S. Shaik, Angew. Chem., Int. Ed., 2008, 47, 3356–3359 CrossRef CAS; (d) S. P. de Visser, L. Tahsini and W. Nam, Chem.–Eur. J., 2009, 15, 5577–5587 CrossRef CAS; (e) S. Fukuzumi, H. Kotani, T. Suenobu, S. Hong, Y.-M. Lee and W. Nam, Chem.–Eur. J., 2010, 16, 354–361 CrossRef CAS; (f) K. A. Prokop, S. P. de Visser and D. P. Goldberg, Angew. Chem., Int. Ed., 2010, 49, 5091–5095 CAS; (g) S. P. de Visser, R. Latifi, L. Tahsini and W. Nam, Chem.–Asian J., 2011, 6, 493–504 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: synthesis and characterization data and kinetic details. CCDC reference numbers 812941. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00386k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2011 |