Enantioselective Mannich reaction of a highly reactive Horner–Wadsworth–Emmons reagent with imines catalyzed by a bifunctional thiourea†
Depeng
Zhao
a,
Dongxu
Yang
a,
Yijie
Wang
a,
Yuan
Wang
a,
Linqing
Wang
a,
Lijuan
Mao
a and
Rui
Wang
*ab
aKey Laboratory of Preclinical Study for New Drugs of Gansu Province, School of Medicine, State Key Laboratory of Applied Organic Chemistry, Institute of Biochemistry and Molecular Biology, Lanzhou University, Lanzhou, 730000, P. R. China. E-mail: wangrui@lzu.edu.cn; Tel: +86-9318912567
bState Key Laboratory of Chiroscience and Department of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong
First published on 13th July 2011
Abstract
N-acyl pyrrole phosphonates were identified as a class of highly reactive HWE reagents. The asymmetric addition of the present HWE reagents to N-carbamoyl imines generated in situ was achieved. The corresponding Mannich adducts can be further transformed to aza-MBH type products in good to excellent enantioselectivities.
Horner–Wadsworth–Emmons (HWE) reagents1 are well known as powerful and frequently used reagents for the construction of α,β-unsaturated carbonyl compounds (Scheme 1). Surprisingly, despite their wide laboratory and industrial applications, there have been few examples concerning their applications in catalytic asymmetric reactions as nucleophiles.2 We guess one of the most critical reasons for their rare studies might be attributed to the relatively low reactivity. Here we report an asymmetric reaction of N-acyl pyrrole phosphonates as highly reactive Horner–Wadsworth–Emmons reagents with N-carbamoyl imines generated in situ and catalyzed by a bifunctional thiourea catalyst. Furthermore, the corresponding adducts can be readily transformed to aza-Morita–Baylis–Hillman (MBH)3,4 type products.
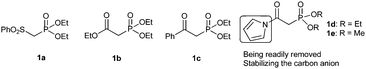 | ||
| Scheme 1 HWE reagents used in the Mannich Reaction. | ||
Although the catalytic asymmetric aza-MBH reaction of imines with α,β-unsaturated ketones or esters is a powerful method to provide chiral functionalized β-amino carbonyl compounds,5 the β-substituted Michael acceptors are generally less reactive under these catalytic systems. Especially for β-aryl substituted α,β-unsaturated esters or ketones, no report has been seen to the best of our knowledge (eqn (1)). In view of this limitation, an alternative approach is needed to obtain chiral aryl substituted aza-MBH products. The present study provides a convenient approach to aryl substituted aza-MBH products through a Mannich/HWE sequence (eqn (2)), and it is complementary to those of existing direct enantioselective aza-MBH methods.
In connection with our continuous interest in hydrogen bonding catalysis6,7 and phosphorus containing nucleophiles,8 we decided to investigate the reaction of HWE reagents with imines. The asymmetric addition of the HWE reagents to imines can provide chiral functionalized phosphonates, which are able to undergo an olefination process with aldehydes or ketones and producing the aza-MBH type products.
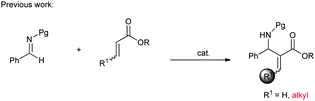 | (1) |
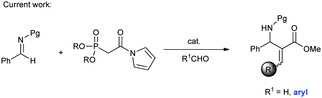 | (2) |
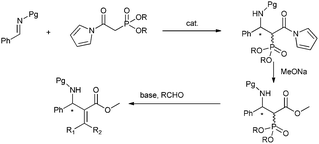
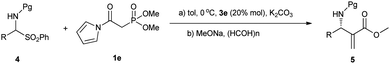
On the basis of the above consideration, we began our investigation with the reaction of N-Boc imine92a with a series of frequently used phosphonates catalyzed by quinine-derived thiourea 3a. The preliminary results were frustrating; it was found that phosphonates 1a, 1b and 1c did not react with N-Boc imine in the presence of a tertiary amine thiourea. Considering the low reactivity of these phosphonates, we then tried to find other base sensitive phosphonates. Then we noticed that N-acyl pyrrole phosphonate 1d might be a promising candidate since 1d was previously used to synthesize α,β-unsaturated compounds promoted by tertiary amine/LiCl10 whereas phosphonates 1a, 1b and 1c generally need metal bases to react with aldehydes. So we proposed the N-acyl pyrrole phosphonate 1d might be reactive enough in the present reaction with the aid of tertiary amine thiourea catalyst. Gratifyingly, when the phosphonate 1d was introduced to the present Mannich reaction, the reaction proceeded smoothly to furnish the desired product. The average ee of the intermediate phosphonate was determined by conversion to aza-MBH product 5aa. As shown in Scheme 2, the N-acyl pyrrole group can be readily transformed to the corresponding methyl ester with MeONa, and the subsequent HWE olefination with paraformaldehyde also occurs at the same conditions. Although the obtained enantioselectivity was proved to be poor (14% ee), we were still encouraged by the high reactivity of 1d and focused on screening other hydrogen bonding catalysts.
 | ||
| Scheme 2 Asymmetric reaction of N-acyl pyrrole phosphonates with imines. | ||
After testing a number of thioureas with diversely structured scaffolds (Scheme 3), we found Takemoto's catalyst 3c gave the best result (59% ee). Further modifications of the tertiary amine group suggested the thiourea 3e bearing a five member ring gave an obviously higher enantioselectivity (75% ee) than 3c. Based on the result, we then tried to optimize the electron-withdrawing group of catalyst through the preparation of derivatives 3g–j. Unfortunately, the enantioselectivities obtained with these catalysts were significantly lower than that provided by 3e. In addition, the monofunctional thioureas 3l, 3m and a squaramide 3k were also proven to be ineffective for the current reaction.
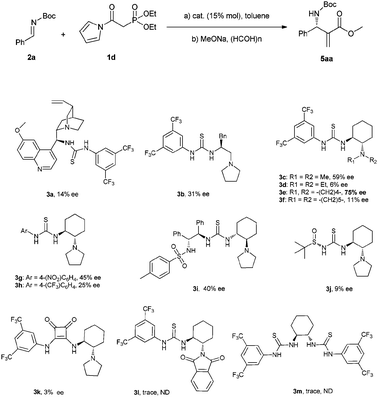 | ||
| Scheme 3 Model reaction and catalysts studied. | ||
Having failed to find a more advantageous catalyst than 3e, we tried other variants such as the substrates. When dimethyl phosphonate 1e was employed instead of 1d, the enantiomeric excess and yield were both slightly increased (78% ee, 82%, Table 1, entry 2). To our delight, the enantiomeric excess increased remarkably to 89% ee when replacing N-Boc imine 2a with N-Cbz imine 2b (Table 1, entry 3). The following solvent screening indicated toluene was the best solvent (Table 1, entries 4–7). By lowering the reaction temperature to 0 °C, the desired adducts can be obtained with 91% ee (entry 8). Further lowering the reaction temperature to −15 °C led to 92% ee without obvious impact on the yield (entry 9). In view of the stability issue of N-Cbz imines, we also attempted to use α-amido sulfones as precursor of in situ generated imines.11 We were pleased to find the reaction also works well in the presence of an aqueous solution of K2CO3 at 0 °C despite 20% mol 3e was required (entry 10).
|
|
||||||
|---|---|---|---|---|---|---|
| Entrya | Pg | 1 | Solvent | T (°C) | Yield (%)b | ee (%)c |
| a Unless otherwise noted, reactions were carried out with 1 (0.375 mmol, 1.5 equiv) and 2 (0.25 mmol) in 4.0 mL toluene for 24 h. b Yield of the isolated product (two steps) after chromatography on silica gel. c The enantiomeric excess was determined by HPLC analysis. d The reaction was performed with in situ generated imine and 20% mol catalyst at 0 °C in 4.0 mL toluene and aqueous K2CO3 (1.5 M, 0.2 mL, 0.3 mmol) for 24 h. | ||||||
| 1 | Boc | 1d | Toluene | r.t. | 79 | 75 |
| 2 | Boc | 1e | Toluene | r.t. | 82 | 78 |
| 3 | Cbz | 1e | Toluene | r.t. | 89 | 89 |
| 4 | Cbz | 1e | THF | r.t. | 65 | 43 |
| 5 | Cbz | 1e | Xylene | r.t. | 73 | 86 |
| 6 | Cbz | 1e | DCM | r.t. | 70 | 51 |
| 7 | Cbz | 1e | CH3CN | r.t. | trace | ND |
| 8 | Cbz | 1e | Toluene | 0 | 87 | 91 |
| 9 | Cbz | 1e | Toluene | −15 | 85 | 92 |
| 10 | Cbz | 1e | Toluene | 0 | 87 | 91d |
Having established two optimal reaction conditions, we chose in situ generated imines to study the scope of this Mannich reaction. As listed in Table 2, good to excellent enantioselectivities were observed for aromatic imines generated in situ regardless of the electronic nature or positions of the substituents. The electronic character of the substrate significantly affected the rate of the Mannich reaction. Electron-rich substrates such as 4h, 4m require longer reaction time to obtain satisfactory conversion than electron-withdrawing ones. For heteroaromatic substrate 4n, employing more sterically hindered 1d instead of 1e led to a significantly higher enantioselectivity (84% vs. 61%, Table 2, entries 19, 20). Good to excellent enantioselectivities were also observed for other heteroaromatic substrates 4o–4q using 1d as the nucleophile. Unfortunately, aliphatic substrates are not quite suitable for the catalysis. Substrates 4r–t were found to be sluggish and gave lower ee's (58–70% ee, Table 2, entries 24–26), while the use of branched substrate 4u resulted in no reaction. In addition, we also attempted to use Boc-protected α-amido sulfones in the present reaction. We were pleased to find they were also applicable to this method despite the enantioselectivities were generally slightly lower than that provided by their Cbz counterparts and longer reaction times were needed.
|
|
||||||
|---|---|---|---|---|---|---|
| Entrya | R | Pg | T (h) | 5 | Yield (%)b | ee (%)c |
| a Unless otherwise noted, reactions were carried out with 1e (0.375 mmol, 1.5 equiv) and 4 (0.25 mmol) in 4.0 mL toluene and aqueous K2CO3 (1.5 M, 0.2 mL, 0.3 mmol) at 0 °C. b Yield of the isolated product (two steps) after chromatography on silica gel. c The enantiomeric excess was determined by HPLC analysis. Absolute configurations of 5a and 5aa were determined to be S and the rest of the products were assigned by analogy with 5a or 5aa. See ESI for details.† d 1d was used as the nucleophile. e Reactions were carried out with 2.0 equiv of 1e at 0 °C for 36 h and rt for another 36 h. f Reactions were carried out at rt. | ||||||
| 1 | C6H5 | Cbz | 24 | 5a | 87 | 91 |
| 2 | C6H5 | Boc | 72 | 5aa | 83 | 86e |
| 3 | 4-FC6H4 | Cbz | 24 | 5b | 87 | 90 |
| 4 | 4-FC6H4 | Boc | 72 | 5ab | 89 | 86e |
| 5 | 4-BrC6H4 | Cbz | 24 | 5c | 87 | 91 |
| 6 | 4-ClC6H4 | Cbz | 24 | 5d | 86 | 86 |
| 7 | 4-ClC6H4 | Boc | 72 | 5ad | 84 | 89e |
| 8 | 3-ClC6H4 | Cbz | 24 | 5e | 80 | 92 |
| 9 | 2-ClC6H4 | Cbz | 24 | 5f | 84 | 91 |
| 10 | 4-MeC6H4 | Cbz | 24 | 5g | 79 | 88 |
| 11 | 4-OMeC6H4 | Cbz | 48 | 5h | 76 | 83 |
| 12 | 3-MeC6H4 | Cbz | 24 | 5i | 87 | 89 |
| 13 | 4-CF3C6H4 | Cbz | 24 | 5j | 81 | 92 |
| 14 | 4-CNC6H4 | Cbz | 24 | 5k | 88 | 93 |
| 15 |
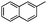
|
Cbz | 24 | 5l | 80 | 87 |
| 16 |
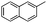
|
Boc | 72 | 5al | 72 | 83e |
| 17 |
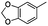
|
Cbz | 48 | 5m | 78 | 96 |
| 18 |
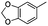
|
Boc | 72 | 5am | 79 | 87e |
| 19 | 2-Furyl | Cbz | 48 | 5n | 76 | 61 |
| 20 | 2-Furyl | Cbz | 48 | 5n | 72 | 84d |
| 21 | 3-Furyl | Cbz | 48 | 5o | 71 | 81d |
| 22 | 2-Thienyl | Cbz | 48 | 5p | 80 | 94d |
| 23 | 3-Thienyl | Cbz | 48 | 5q | 85 | 86d |
| 24 | PhCH2CH2 | Cbz | 96 | 5r | 65 | 63f |
| 25 | Pr | Cbz | 96 | 5s | 69 | 70f |
| 26 | iBu | Cbz | 96 | 5t | 57 | 57f |
| 27 | iPr | Cbz | 72 | 5u | NR | NDf |
To illustrate the utility of our reaction products, we also investigated the olefination of phosphonates 6a or 9a with other aldehydes. When MeONa was used as a base, Z products were found to be dominant for both benzaldehyde and cinnamaldehyde. Whereas in the case of a proazaphosphatrane P(i-BuNCH2CH2)3N12 serving as a base, we were surprised to find the E isomer was the major product. In order to verify the generality of this phenomenon, we then examined other addition products formed in Table 2. The results listed in Scheme 4 indicated all reactants investigated including aryl, heteroaryl and alkyl 6 and 9 complied with the rule aforementioned with E/Z up to 10.4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. So, with this versatile Mannich product, we could provide either E or Z aza-MBH type product respectively with specific bases. The stereochemistry of the resulting products was assigned on the basis of the chemical shift values of vinyl protons in the 1H NMR and NOESY analyses.13
:1. So, with this versatile Mannich product, we could provide either E or Z aza-MBH type product respectively with specific bases. The stereochemistry of the resulting products was assigned on the basis of the chemical shift values of vinyl protons in the 1H NMR and NOESY analyses.13
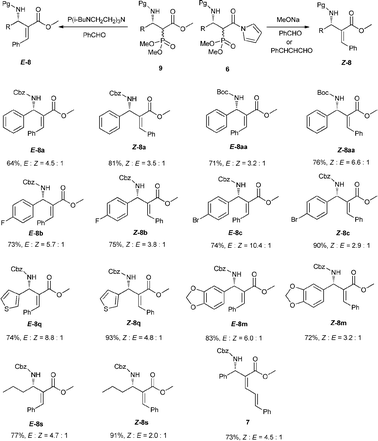 | ||
| Scheme 4 Transformations of the corresponding Mannich products. | ||
In summary, we have identified N-acyl pyrrole phosphonates as a class of highly reactive nucleophiles, and achieved the asymmetric addition of the present HWE reagent to N-carbamoyl imines generated in situ. The corresponding Mannich adducts were further transformed to aza-MBH products in good to excellent enantioselectivities. More importantly, the Mannich adducts can also be converted to aryl substituted aza-MBH products, in which access to both E/Z isomers is achieved by a switch in the base source. Further investigations on the reaction scope of the N-acyl pyrrole phosphonates and application of this strategy in peptide modifications are in progress.
We gratefully acknowledge financial support from NSFC (20932003, and 90813012) and the National S&T Major Project of China (2009ZX09503-017).
Notes and references
- (a) W. S. Wadsworth and W. D. Emmons, J. Am. Chem. Soc., 1961, 83, 1733 CrossRef CAS; (b) B. E. Maryanoff and A. B. Reize, Chem. Rev., 1989, 89, 863 CrossRef CAS.
- (a) T. Arai, H. Sasai, K. Yamaguchi and M. Shibasaki, J. Am. Chem. Soc., 1998, 120, 441 CrossRef CAS; (b) T. S. Pham, L. Balázs, I. Petneházy and Z. Jászay, Tetrahedron: Asymmetry, 2010, 21, 346–351 CrossRef CASFor an Horner reagent, see: (c) S. Mazzotta, L. Gramigna, L. Bernardi and A. Ricci, Org. Process Res. Dev., 2010, 14, 687–691 CrossRef CAS. For other phosphonate-containing nucleophile involved in organocatalyzed enantioselective Mannich reactions, see: (d) J. C. Wilt, M. Pink and J. N. Johnston, Chem. Commun., 2008, 4177 RSC; (e) R. D. Momo, F. Fini, L. Bernardi and A. Ricci, Adv. Synth. Catal., 2009, 351, 2283 CrossRef CAS and references therein.
- For recent review on the MBH and aza-MBH reactions, see: (a) D. Basavaiah, B. S. Reddy and S. S. Badsara, Chem. Rev., 2010, 110, 5447 CrossRef CAS; (b) V. Declerck, J. Martinez and F. Lamaty, Chem. Rev., 2009, 109, 1 CrossRef CAS; (c) G. Masson, C. Housseman and J. Zhu, Angew. Chem., Int. Ed., 2007, 46, 4614 CrossRef CAS; (d) Y.-L. Shi and M. Shi, Eur. J. Org. Chem., 2007, 290 Search PubMed.
- For unconventional methods to access aza-MBH type reaction products, see: (a) Y. Zhang, Y.-K. Liu, T.-R. Kang, Z.-K. Hu and Y.-C. Chen, J. Am. Chem. Soc., 2008, 130, 2456 CrossRef CAS; (b) C. Cassani, L. Bernardi, F. Fini and A. Ricci, Angew. Chem., Int. Ed., 2009, 48, 5694 CrossRef CAS; (c) T. E. Reynolds and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 7806 CrossRef CAS; (d) B. M. Trost and C. K. Chung, J. Am. Chem. Soc., 2006, 128, 10358 CrossRef CAS; (e) A. Yamaguchi, N. Aoyama, S. Matsunaga and M. Shibasaki, Org. Lett., 2007, 9, 3387 CrossRef CAS.
- For selected recent examples of the catalytic asymmetric aza-MBH reaction, see: (a) M. Shi and Y.-M. Xu, Angew. Chem., Int. Ed., 2002, 41, 4507 CrossRef CAS; (b) K. Matsui, S. Takizawa and H. Sasai, J. Am. Chem. Soc., 2005, 127, 3680 CrossRef CAS; (c) M. Shi, L.-H. Chen and C.-Q. Li, J. Am. Chem. Soc., 2005, 127, 3790 CrossRef CAS; (d) N. Abermil, G. Masson and J. Zhu, Org. Lett., 2009, 11, 4648 CrossRef CAS; (e) F. Zhong, Y. Wang, X. Han, K. Huang and Y. Lu, Org. Lett., 2011, 13, 1310 CrossRef CAS and references therein.
- For selected examples of thiourea catalyzed reactions from our group, see: (a) X. Jiang, Y. Cao, Y. Wang, L. Liu, F. Shen and R. Wang, J. Am. Chem. Soc., 2010, 132, 15328 CrossRef CAS; (b) X. Jiang, D. Fu, G. Zhang, Y. Cao, L. Liu, J. Song and R. Wang, Chem. Commun., 2010, 46, 4294 RSC; (c) X. Jiang, G. Zhang, D. Fu, Y. Cao, M. F. Shen and R. Wang, Org. Lett., 2010, 12, 1544 CrossRef CAS; (d) X. Liu, L. Deng, X. Jiang, W. Yan, C. Liu and R. Wang, Org. Lett., 2010, 12, 876 CrossRef CAS.
- For selected examples on thiourea catalysis, see: (a) M. S. Sigman and E. N. Jacobsen, J. Am. Chem. Soc., 1998, 120, 4901 CrossRef CAS; (b) A. Berkessel, F. Cleemann and S. Mukherjee, Angew. Chem., Int. Ed., 2005, 44, 7466 CrossRef CAS; (c) Y. Yamaoka, H. Miyabe and Y. Takemoto, J. Am. Chem. Soc., 2007, 129, 6686 CrossRef CAS; (d) N. J. A. Martin, L. Ozores and B. List, J. Am. Chem. Soc., 2007, 129, 8976 CrossRef CAS; (e) Y. Sohtome, Y. Hashimoto and K. Nagasawa, Adv. Synth. Catal., 2005, 347, 1643 CrossRef CAS; (f) R. R. Knowles, S. Lin and E. N. Jacobsen, J. Am. Chem. Soc., 2010, 132, 5030 CrossRef CAS; (g) T. Bui, S. Syed and C. F. Barbas III, J. Am. Chem. Soc., 2009, 131, 8758 CrossRef CAS; (h) J. Wang, H. Li, L. Z. Zu, H. X. Xie, W. H. Duan and W. Wang, J. Am. Chem. Soc., 2006, 128, 12652 CrossRef CAS; (i) J. Ye, D. J. Dixon and P. Hynes, Chem. Commun., 2005,(35), 4481 RSC; (j) C. Wang, X. Dong, Z. Zhang, Z. Xue and H. Teng, J. Am. Chem. Soc., 2008, 130, 8606 CrossRef CAS.
- (a) D. Zhao, Y. Yuan, A. S. C. Chan and R. Wang, Chem.–Eur. J., 2009, 15, 2738 CrossRef CAS; (b) D. Zhao, Y. Wang, L. Mao and R. Wang, Chem.–Eur. J., 2009, 15, 10983 CrossRef CAS; (c) D. Zhao, L. Mao, Y. Wang, D. Yang, Q. Zhang and R. Wang, Org. Lett., 2010, 12, 1880 CrossRef CAS; (d) D. Zhao, L. Mao, D. Yang and R. Wang, J. Org. Chem., 2010, 75, 6756 CrossRef CAS; (e) L. Hong, W. Sun, C. Liu, D. Zhao and R. Wang, Chem. Commun., 2010, 46, 2856 RSC; (f) W. Sun, L. Hong, C. Liu and R. Wang, Org. Lett., 2010, 12, 3914–3917 CrossRef CAS.
- For organocatalyzed Mannich reaction of N-Cbz and N-Boc imines with malonates, see: (a) T. B. Poulsen, C. Alemparte, S. Saaby, M. Bella and K. A. Jørgensen, Angew. Chem., 2005, 117, 2956 CrossRef; (b) S. Lou, B. Taoka, A. Ting and S. E. Schaus, J. Am. Chem. Soc., 2005, 127, 11256 CrossRef CAS; (c) A. Ting, S. Lou and S. E. Schaus, Org. Lett., 2006, 8, 2003 CrossRef CAS; (d) C. M. Bode, A. Ting and S. E. Schaus, Tetrahedron, 2006, 62, 11499 CrossRef CAS; (e) A. L. Tillman, J. Ye and D. J. Dixon, Chem. Commun., 2006, 1191 RSC; (f) J. Song, Y. Wang and L. Deng, J. Am. Chem. Soc., 2006, 128, 6048 CrossRef CAS; (g) T.-Y. Liu, H.-L. Cui, J. Long, B.-J. Li, Y. Wu, L.-S. Ding and Y.-C. Chen, J. Am. Chem. Soc., 2007, 129, 1878 CrossRef CAS; (h) J. Song, H.-W. Shih and L. Deng, Org. Lett., 2007, 9, 603 CrossRef CAS; (i) F. Fini, L. Bernardi, R. P. Herrera, D. Pettersen, A. Ricci and V. Sgarzani, Adv. Synth. Catal., 2006, 348, 2043 CrossRef CAS; (j) B. Niess and K. A. Jørgensen, Chem. Commun., 2007, 1620 RSC; (k) O. Marianacci, G. Micheletti, L. Bernardi, F. Fini, M. Fochi, D. Pettersen, V. Sgarzani and A. Ricci, Chem.–Eur. J., 2007, 13, 8338 CrossRef CAS.
- N. Yamagiwa, H. Qin, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2005, 127, 13419 CrossRef CAS.
- For examples of α-amido sulfones as imine precursors in Mannich type reactions, see: (a) H. Zhang, S. Syed and C. F. Barbas III, Org. Lett., 2010, 12, 708 CrossRef CAS; (b) J. Song, H. W. Shih and L. Deng, Org. Lett., 2007, 9, 603 CrossRef CAS; (c) F. Fini, V. Sgarzani, D. Pettersen, R. P. Herrera, L. Bernardi and A. Ricci, Angew. Chem., Int. Ed., 2005, 44, 7975 CrossRef CAS.
- V. R. Chintareddy, A. Ellern and J. G. Verkade, J. Org. Chem., 2010, 75, 7166 CrossRef CAS.
- For details, see the ESI†.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical data (1H NMR, 13C NMR and HRMS) for all new compounds. See DOI: 10.1039/c1sc00351h |
| This journal is © The Royal Society of Chemistry 2011 |