Visualising the hypoxia selectivity of cobalt(III) prodrugs†
Byung J.
Kim
,
Trevor W.
Hambley
and
Nicole S.
Bryce
*
School of Chemistry, University of Sydney, Sydney, Australia. E-mail: nicole.bryce@sydney.edu.au; Fax: +61 2 9351 3329; Tel: +61 2 9351 4233
First published on 11th August 2011
Abstract
Hypoxic cancer cells have a more aggressive phenotype than cells in a normoxic environment and are often refractory to current chemotherapy protocols due to an altered proliferation rate. They also represent a means of targeting cancer chemotherapy. We have developed a 3-dimensional cell culture model that readily identifies live hypoxic cells by expression of the Eos photo-convertible green fluorescent protein. Using this model, we have examined the diffusion and hypoxia selectivity of two model Co(III) based compounds. The Co(III) compounds have been designed to target hypoxic cells within a tumour and have fluorescent axial ligands. The fluorescent ligands function as model cytotoxins and their fluorescence is quenched on binding to Co(III). The ligands are released upon the reduction of Co(III) to Co(II) and the recovery of fluorescence enables the detection of the released ligands by confocal microscopy. Despite similarities in electrochemical potential and cytotoxicity, the penetration of the compounds into spheroids and uptake into hypoxic cells was found to differ depending on the properties of the released ligand.
Introduction
Hypoxia occurs in regions of a solid tumour that have been deprived of adequate oxygen supply1 as a result of temporary disruptions in blood flow or because of an increased diffusion distance from the microvasculature (>70 μm).2,3 Hypoxic cancer cells have previously been shown to have altered gene expressions resulting from the activation of the basic helix-loop-helix transcription factor HIF1, leading to more aggressive phenotypes and a poor prognosis for the patient.4–9 Chemo-resistant and radio-resistant cancer cell populations have been associated with the hypoxic regions of cancers and the diffusion of drugs into these regions is limited by the same factors that limit the diffusion of O2.10,11 Hypoxic tumour cells often have a lower proliferation rate than cells in the normoxic areas of the tumour which limits the effects of anti-cancer drugs that target the cell cycle and/or rely on rapid proliferation.12 Successful drug delivery to hypoxic areas of solid tumours is therefore of vital importance in the effective treatment of cancer by chemotherapy.Drug diffusion into solid tumours is influenced by the charge, lipophilicity and solubility of the compound13 as well as factors such as extracellular matrix composition and P-glycoprotein status.14 Diffusion of fluorescent Pt based compounds into multicellular tumour spheroids has shown that the outer layers of the spheroid receive or accumulate a much higher dose of drug than the cells in the centre.15 This leaves a population of hypoxic cells in the centre of the spheroid that potentially do not receive an adequate dose of the anti-cancer compound.
Previous prodrug design strategies have utilized the preferential reduction of Co(III) to Co(II) and the consequent release of cytotoxic ligands when the compound is in an environment that leads to reduction such as in hypoxia.16–19Co(III) is kinetically inert whereas Co(II) is quite labile and undergoes rapid ligand exchange with water.20 Hence, under hypoxic conditions, the complexes are reduced to Co(II) complexes which rapidly undergo aquation in cells, releasing the cytotoxic agent, whereas under normoxic conditions, the Co(II) is rapidly re-oxidized.21 The ligands of the cobalt complexes can be modified to improve the physicochemical characteristics of these complexes to tailor them for hypoxia selectivity.17 Properties such as the cytotoxicity, potential of the Co(III)/Co(II) couple, overall charge, solubility, and lipophilicity can be manipulated.22 An additional useful property of Co(III) is that upon complexation, the inherent fluorescence of some fluorophores is quenched.23,24 This allows monitoring of ligand release in cellsvia the return of fluorescence25,26 and the rational optimization of the pharmacological properties of the prodrug.
The aims of this project were to develop a 3-dimensional system of fluorescently labelled hypoxic cells within an avascular solid tumour model and determine if Co(III) prodrugs effectively target the hypoxic regions of the model. Two Co(III) complexes, [Co(C343)2(cyclam)]Cl (1) and [Co(AQ2C)2(cyclam)]Cl (2), were synthesized with the fluorescent ligands anthraquinone-2-carboxylic acid (AQ2CH) or coumarin-343 (C343H) occupying the axial sites and the tetradentate ligand 1,4,8,11-tetraazacyclotetradecane (cyclam) in the equatorial sites. To determine whether these cobalt complexes are able to penetrate deep into a tumour and preferentially release the fluorescent ligands in hypoxic conditions, we generated a DLD-1 colon carcinoma cell line expressing the photo-convertible EosFP green fluorescent protein under the control of the hypoxia response element (HRE). These cells were then grown as multicellular spheroids and activation of the reporter gene in response to hypoxic conditions was observed in the core of the spheroids. By monitoring the fluorescence of the released ligand and of the hypoxic cells we have shown that Co(III) complexes are effective at releasing the fluorescent ligands within the hypoxic regions of the tumor model.
Results and discussion
Characterization of [Co(AQ2C)2(cyclam)]Cl
Cobalt complexes are promising hypoxia activated prodrugs due to the marked difference in lability between the Co(III) and Co(II) oxidation states.16 Two cobalt complexes were investigated in this study, each having a cyclam ligand occupying the four equatorial sites and two carboxylate donor groups occupying the axial sites. Two fluorescent compounds were chosen as axial ligands in this study, anthraquinone-2-carboxylic acid (AQ2CH) and Coumarin-343 (C343H). Fig. 1 shows the chemical structure of the Co(III) compounds that were synthesized in this study, [Co(AQ2C)2(cyclam)]Cl (1) and [Co(C343)2(cyclam)]Cl (2). The synthesis and characterization of [Co(C343)2(cyclam)]ClO4 have been previously described.27![Compounds generated and used in this study. (1) [Co(AQ2C)2(cyclam)]Cl, (2) [Co(C343)2(cyclam)]Cl.](/image/article/2011/SC/c1sc00337b/c1sc00337b-f1.gif) | ||
| Fig. 1 Compounds generated and used in this study. (1) [Co(AQ2C)2(cyclam)]Cl, (2) [Co(C343)2(cyclam)]Cl. | ||
Characterization of [Co(AQ2C)2(cyclam)]Cl was performed using NMR, IR, MS, and CHN elemental analysis. The 1H and 13C NMR peaks were too broad to be able to assign shifts, possibly due to the conformational dynamics of the cyclam. This could also be due to the fluorophore causing a pseudo-paramagnetic character as suggested by Yamamoto and colleagues for related C343ha complexes.27 The MS showed that the complex was successfully synthesized, showing the parent ion as well as a fragment ion without the AQ2CH ligands. CHN elemental analysis showed an acceptable C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H:N ratio which was complemented by the MS. The structure of the complex is proposed to be octahedral with AQ2CH coordinated in a monodentate fashion through the carboxylate ion and cyclam adopting a trans geometry, occupying the other four coordination sites.
:H:N ratio which was complemented by the MS. The structure of the complex is proposed to be octahedral with AQ2CH coordinated in a monodentate fashion through the carboxylate ion and cyclam adopting a trans geometry, occupying the other four coordination sites.
Fluorescence studies
The excitation and emission spectra of [Co(AQ2C)2(cyclam)]Cl were measured and compared to those of the free ligand, AQ2CH (Supplementary Fig. 1A and 1B†). Upon coordination to cobalt, the fluorescence of the AQ2CH ligand was quenched to 21% of that observed for the equivalent concentration of AQ2CH. The excitation and emission spectra of [Co(C343)2(cyclam)]Cl have been previously measured and compared to those of the free ligand, C343H.27The effect of reductants on the fluorescence of [Co(AQ2C)2(cyclam)]Cl
The fluorescence of a DMA solution of [Co(AQ2C)2(cyclam)]Cl was monitored over 24 h after the addition of a 10 fold excess of the cellular reductant sodium ascorbate (Fig. 2). Sodium ascorbate may reduce the complex allowing for the rapid aquation of the complex, or alternatively coordinate to the cobalt complex thus displacing the ligand. Fluorescence appears almost immediately after the addition of sodium ascorbate in deoxygenated conditions, indicated by the steep increase in the curve. The fluorescence intensity starts to decrease gradually, indicating that the limit on the effects of ascorbate on the cobalt complex was reached. This is in contrast to the rate of reduction of the [Co(AQ2C)2(cyclam)]Cl solution in oxygenated conditions where, following the addition of sodium ascorbate, the fluorescence intensity increases at a slower rate, indicative of a slower increase in the concentration of fluorophore being displaced from the cobalt complex. The [Co(AQ2C)2(cyclam)]Cl therefore is preferentially reduced in a hypoxia selective manner and the difference is consistent with reoxidation competing with ligand release in the oxygenated solution.21![Reduction of [Co(AQ2C)2(cyclam)]Cl in oxygenated and deoxygenated conditions. Maximum fluorescence emission of [Co(AQ2C)2(cyclam)]Cl over 24 h after addition of sodium ascorbate in oxygenated and deoxygenated conditions. The fluorescence intensity has been normalized to 100.](/image/article/2011/SC/c1sc00337b/c1sc00337b-f2.gif) | ||
| Fig. 2 Reduction of [Co(AQ2C)2(cyclam)]Cl in oxygenated and deoxygenated conditions. Maximum fluorescence emission of [Co(AQ2C)2(cyclam)]Cl over 24 h after addition of sodium ascorbate in oxygenated and deoxygenated conditions. The fluorescence intensity has been normalized to 100. | ||
Cytotoxicity studies
The cytotoxicity of C343H, AQ2CH, and their Co(cyclam) complexes were determined using the MTT assay in both normoxic and hypoxic conditions. The concentrations of ligands and complexes which resulted in 50% cell viability (IC50) after 72 h incubation are shown in Table 1. The results for both normoxic and hypoxic environments were similar for all compounds tested. Both cobalt complexes showed a high degree of insolubility in water so concentrated samples were prepared to ensure the solvent systems were not toxic to the DLD-1 cells.| Compound | Normoxia | Hypoxia |
|---|---|---|
| [CoCl2(cyclam)]Cl | >200 | >200 |
| C343H | 99 ± 4 | >200 |
| [Co(C343)2(cyclam)]Cl | 1.3 ± 0.7 | 1.9 ± 0.2 |
| AQ2CH | >200 | 177 ± 0.2 |
| [Co(AQ2C)2(cyclam)]Cl | 35.6 ± 0.9 | 20.4 ± 0.1 |
[Co(C343)2(cyclam)]Cl is considerably more cytotoxic than either of the components, C343H and [CoCl2(cyclam)]Cl, as previously found by Yamamoto et al.27 using the A2780 (ovarian carcinoma) cell line. The compounds were tested on DLD-1 cells in this study instead of A2780 cells due to the difficulty associated with forming A2780 spheroids, an important consideration when testing the possible accumulation of cobalt complexes in hypoxic regions.
The same trend holds for the [Co(AQ2C)2(cyclam)]Cl complex as it has greater cellular cytotoxicity than the ligand or the cobalt starting material. The AQ2CH may be cytotoxic through its reversible DNA intercalation,28,29 but the ligand itself is hypoxia selective due to the generation of damaging anion radical intermediates by the reduction of the quinone moiety.30,31[Co(AQ2C)2(cyclam)]Cl, is 10–27-fold less cytotoxic than [Co(C343)2(cyclam)]Cl in hypoxic and normoxic systems respectively. This may be associated with the rapid release of AQ2CH observed in the reduction experiments, resulting in reduced levels of the prodrug that is able to modify the delivery of the AQ2CH.
Electrochemical studies
It is believed that hypoxia activated prodrugs should ideally have a reduction potential in the region of −0.1 to −0.5 V (vs.NHE) for activation by common flavoproteins and for successful redox cycling.32 The reduction potentials, and reversibility of the reduction of the complexes, [CoCl2(cyclam)]Cl, [Co(C343)2(cyclam)]Cl and [Co(AQ2C)2(cyclam)]Cl were examined using cyclic voltammetry. Table 2 reveals that the reduction potentials of [Co(C343)2(cyclam)]Cl and [Co(AQ2C)2(cyclam)]Cl are more negative than that of the starting material [CoCl2(cyclam)]Cl. This is probably due to the electron donating ability of the negatively charged oxygen donors of C343H and AQ2CH that stabilizes the 3+ oxidation state. The reduction is a one electron process as delta E of the Co(III/II) couple is comparable to the delta E of the fc/fc+ couple and the reduction is also reversible as ipc/ipa is close to 1. The AQ2C ligand was shown to be redox active and the corresponding cobalt complex had a similar cyclic voltammogram with an additional peak associated with the redox active Co(III/II) metal centre. The coordination of the AQ2CH ligand through the carboxylate results in a shift in the peaks associated with one of the electrochemical reactions of the free ligand.33| Complexes | E° vs. fc/fc+ (mV) | E° vs. NHE (mV) | ΔECo(III/II) (mV) |
|---|---|---|---|
| [CoCl2(cyclam)]Cl | −845 | −125 | 73 |
| [Co(C343)2(cyclam)]Cl | −1107 | −387 | 72 |
| [Co(AQ2C)2(cyclam)]Cl | −970 | −250 | 63 |
Generation of a model system to visualize hypoxic cells in 3-dimensions
The second aim of the project was to create a model system of fluorescently labelled hypoxic cells that could form 3-dimensional spheroids. Non-fluorescent HRE-reporter systems have previously been successfully used to identify compounds that directly inhibit expression of genes that are regulated by the HRE element,35 but such systems do not reveal whether a compound is able to successfully target hypoxic cells deep within a tumour. Spheroids mimic the avascular stages of solid tumours in vivo.36 The cells on the periphery of the spheroid actively proliferate, similar to cells near blood vessels, while the hypoxic cells in the centre are quiescent, like those distant from blood vessels.37To achieve this, four copies of the HRE element from the human vascular endothelial growth factor (VEGF) gene were inserted upstream of a minimal CMV promoter38 driving the expression of the photo-convertible green-red fluorescent protein EosFP. DLD-1 human colon carcinoma cells were stably transfected and a polyclonal cell line generated (HRE-Eos). Cells grown in normoxic conditions did not express detectable levels of EosFP by fluorescent microscopy (Fig. 3A), whereas EosFP was detected in those cells incubated in 0.5% O2 for 48 h indicating that the expression of the fluorescent protein was specific to hypoxic conditions (Fig. 3B). 24 h treatment with the iron chelator desferroxamine (Fig. 3C) also induced expression of EosFP via the HIF-1 pathway. HRE-Eos cells were grown as tumour spheroids and the cells in the centre of the spheroid fluoresced green indicating the generation of a hypoxic environment (Fig. 3G–I).
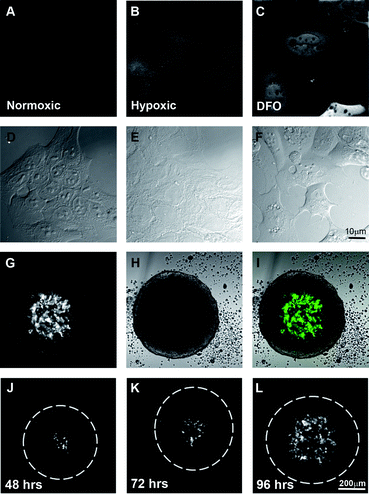 | ||
| Fig. 3 Hypoxia-dependent expression of EosFP in an artificially hypoxic environment as well as in the centre of spheroid cultures. Widefield fluorescent images of DLD-1 cells expressing HRE-EosFP grown in 2-dimensional culture in A. normoxia, B. hypoxia or C. normoxia plus desferroxamine with their respective DIC images D–F. Single confocal images indicating that HRE-EosFP is expressed in the central region of spheroids G–I. Time course of development of hypoxic areas in spheroids as shown by single confocal images of the centre of the spheroid at J. 48, K. 72 and L. 96 h. Scale bar 10 μm for A–F and 200 μm for G–L. | ||
Spheroids were examined daily (Fig. 3J–L), with hypoxia evident after 48 h in a small number of cells in the centre of the spheroid and by day 4, green fluorescent cells were routinely detected throughout the central region of the spheroid and up to 80–100 μm from the outer edge of the spheroid (Fig. 3L) as expected based on the known limitation of oxygen diffusion.2,3 Spheroids can also be grown using various techniques to specific, highly reproducible sizes in large numbers, making them suitable for use in drug screens.39–41 Thus we believe that the HRE-Eos cells grown in 3-dimensional culture represent an excellent model for visualizing fluorescent drug distribution/targeting in hypoxic conditions.
Distribution of the fluorescent ligands in spheroids
After 3 days of culture, the spheroids were dosed with 10 μM of either ligand or Co complex for 24 h and then examined by confocal microscopy. AQ2CH alone is only able to penetrate the spheroid to a depth of ∼70–80 μm (Fig. 4A), which is similar to what is seen for the diffusion of oxygen,13doxorubicin and the Pt complex Pt1C3.15 The merged image (Fig. 4C), shows that the fluorescence of AQ2CH does not overlap with that of the green hypoxic cells (Fig. 4B). [Co(AQ2C)2(cyclam)]Cl is able to penetrate further into the spheroid and release the AQ2CH ligand as shown by the presence and increase in fluorescence at a distance of 100–200 μm into the spheroid (Fig. 4D) compared to AQ2CH alone (Fig. 4A), but there is little co-localization or targeting seen in the merged image (Fig. 4F) of the released AQ2CH ligand with the green hypoxic cells (Fig. 4E) in the centre of the spheroid.![Localization of fluorescent ligands in 3-dimensional culture of DLD-1 cells expressing HRE-EosFP. Single confocal images of the centre of the spheroid are shown. A. AQ2CH. B. HRE-EosFP. C. Merge of A (blue) and B (green). D. [Co(AQ2C)2(cyclam)]Cl, E. HRE-EosFP, F. Merge of D (blue) and E (green). G. C343H. H. Green fluorescence from C343H. I. Merge of G (blue) and H (green). J. [Co(C343)2(cyclam)]Cl, K. Green fluorescence from C343H, L. Merge of J (blue) and K (green). Scale bar 200 μm.](/image/article/2011/SC/c1sc00337b/c1sc00337b-f4.gif) | ||
| Fig. 4 Localization of fluorescent ligands in 3-dimensional culture of DLD-1 cells expressing HRE-EosFP. Single confocal images of the centre of the spheroid are shown. A. AQ2CH. B. HRE-EosFP. C. Merge of A (blue) and B (green). D. [Co(AQ2C)2(cyclam)]Cl, E. HRE-EosFP, F. Merge of D (blue) and E (green). G. C343H. H. Green fluorescence from C343H. I. Merge of G (blue) and H (green). J. [Co(C343)2(cyclam)]Cl, K. Green fluorescence from C343H, L. Merge of J (blue) and K (green). Scale bar 200 μm. | ||
C343H (Fig. 4G) penetrates slightly further into the spheroid than AQ2CH (Fig. 4A), showing a similar distribution profile to that of [Co(AQ2C)2(cyclam)]Cl (Fig. 4D). We were unable to detect the presence of hypoxic cells as the fluorescence of the C343H overlapped with the fluorescence of the Eos protein (Fig. 4H). The green fluorescence of C343H was able to be detected throughout the spheroid with the exception of a central region of ∼150 μm diameter, indicating that the C343H ligand is able to penetrate throughout the majority of the spheroid.
Treatment of the spheroids with the [Co(C343)2(cyclam)]Cl results in the release of the C343H ligand in a different distribution profile (Fig. 4J) to that seen with C343H alone (Fig. 4G). While the green fluorescence distribution profile remains the same, (Fig. 4K cf. 4H) the blue fluorescence profile shows the development of a region of fluorescence that has accumulated in the centre of the spheroid (Fig. 4J). This result shows that the [Co(C343)2(cyclam)]Cl is able to deliver the C343H ligand to the central, hypoxic region of the spheroid; however, due to the green fluorescence of the C343H ligand, we cannot confirm whether the C343H is accumulating within hypoxic cells or within the hypoxic region of the spheroid.
Photo-conversion of the Eos protein
Due to the overlapping fluorescence spectra of C343H and EosFP, we utilized the unique photo-conversion properties of the EosFP protein. The use of the photo-convertible EosFP as a fluorescent marker, instead of a monochromatic fluorescent protein such as mCherry or GFP,42,43 allows for greater flexibility in the available fluorescent spectrum, an important consideration when working with the inherent fluorescence of DNA intercalators such as the coumarins and anthraquinones. Normally, EosFP emits only green fluorescence (Fig. 5A–C). However, after exposure to UV light, EosFP is photo-converted to a red fluorescent protein (Fig. 5D–F), with minimal remaining green fluorescence44 (Fig. 5D). The photo-conversion process is very efficient and all cells within the centre of the spheroid are able to be photo-converted as shown by the yellow cells in the merge (Fig. 5F, magnified in Fig. 5G), of the green (Fig. 5A), and the red (Fig. 5E), before and after photo-conversion.![Identification of cells in hypoxic areas after green to red photo-conversion of HRE-Eos within spheroids. DLD-1 cells expressing HRE-Eos were grown as spheroids and imaged viaconfocal microscopy using standard A. FITC and B. Texas Red parameters. C. Merge of A and a brightfield image of the spheroid. After exposure to UV light, the same spheroid was imaged with standard D. FITC and E. Texas Red parameters to show photo-conversion. F. Merge of images A (green) and E (red). Spheroids treated with [Co(C343)2(cyclam)]Cl and exposed to UV light were imaged with standard H. UV, I. FITC and J. Texas Red parameters. The region marked in I is magnified in K–O. Scale bar 200 μm.](/image/article/2011/SC/c1sc00337b/c1sc00337b-f5.gif) | ||
| Fig. 5 Identification of cells in hypoxic areas after green to red photo-conversion of HRE-Eos within spheroids. DLD-1 cells expressing HRE-Eos were grown as spheroids and imaged viaconfocal microscopy using standard A. FITC and B. Texas Red parameters. C. Merge of A and a brightfield image of the spheroid. After exposure to UV light, the same spheroid was imaged with standard D. FITC and E. Texas Red parameters to show photo-conversion. F. Merge of images A (green) and E (red). Spheroids treated with [Co(C343)2(cyclam)]Cl and exposed to UV light were imaged with standard H. UV, I. FITC and J. Texas Red parameters. The region marked in I is magnified in K–O. Scale bar 200 μm. | ||
Re-examination of the [Co(C343)2(cyclam)]Cl treated spheroids with photo-converted EosFP (Fig. 5H–O), revealed a distinct difference in the localization of the hypoxic cells in [Co(C343)2(cyclam)]Cl treated spheroids (Fig. 5J, N) compared to untreated spheroids (Fig. 5E). Diminished red fluorescence was seen within the central 150 μm of the treated spheroid (Fig. 5J, N) that correlated with the increased blue fluorescence of the released C343H ligand (Fig. 5L). Fig. 5L also shows the localization of the C343H ligand fluorescence into punctuate spots, indicative of cellular accumulation. The loss of red fluorescence is consistent with cell deathvia the targeted accumulation of the C343H ligand into hypoxic cells in the central region of the spheroid.
The differences seen between the biological behaviour of the two Co complexes is surprising, as the compounds are expected to have similar stabilities and rates of reduction. The most significant difference lies in the presence of the protonatable amine group on C343H. At physiological pH we expect this group to be protonated to only a very small extent. The release of C343H from the Co(III) moiety is not completely hypoxia selective as there is a band of bright fluorescence at the edge of the spheroid. However, the delayed release of the ligands in response to cellular reductants compared to AQ2CH allowed for more of the [Co(C343)2(cyclam)]Cl to penetrate into the spheroid and be reduced in the hypoxic environment. This is of great significance as we can utilize the relationship between reduction potential and fluorescent ligand release of a compound to design other effective hypoxia activated cytotoxic compounds that penetrate deep within the tumour.
Conclusions
We report here the generation of a live model of an avascular tumour with easily detectable, dual colour hypoxic cells that is able to be generated without specialised hypoxic chambers. By using a hypoxia activated fluorescent marker, we have generated a system that can be readily used to screen compounds for identifying hypoxia localization and/or activity in 3D in living cells. Two model cobalt compounds were synthesised and despite having similar reduction potentials, the release of the fluorescent ligands within the tumour model differed significantly. An important consideration in future compound design will be the balance of tumour penetration, hypoxic cell targeting, ligand release and ligand uptake in hypoxic cells, which can be monitored using this model system. New anti-cancer compounds which are designed to specifically target the hypoxic cells in a tumour will be of vital importance to lessen the possibility of metastasis and the development of drug resistance in cells that may remain in a tumour after treatment with current protocols.Experimental procedures
Chemical syntheses
Synthesis of trans-[CoCl2(cyclam)]Cl45 was as previously described.Preparation of [Co(C343)2(cyclam)]Cl
Due to the light sensitivity of coumarin-343, all steps in the following procedure were carried out in the dark.[Co(C343)2(cyclam)]Cl was synthesised using a modification of a previous procedure.25C343H (0.24 g, 0.84 mmol) was suspended in MeOH (30 ml), previously dried with Na2SO4, and triethylamine was added (151 μl, 0.84 mmol). The mixture was stirred for 10 min before the addition of [CoCl2(cyclam)]Cl (0.10 g, 0.27 mmol) as a solid, and the solution was further stirred for 3 days. The unreacted C343H was filtered off and the product was purified by Dowex anion exchange column chromatography (1 × 8, 200/400). The Dowex was previously charged with 1 M HCl and then flushed with water until reaching pH 7. The column was then equilibrated with dry MeOH. The product was eluted with MeOH until the resulting solution was colourless and the solvent was evaporated in vacuo. The bright orange solids were filtered and the solid washed with cold water (30 ml), ethyl acetate (20 ml) and diethyl ether (10 ml) to yield the product as a bright orange solid. (yield: 0.17 g. 0.20 mmol, 72% yield). Anal. Calc. for [Co(C16H14NO4)2(C10H24N4)]Cl: C 58.43, H 6.07, N 9.73. Found C 58.32, H 6.04, N 9.51. IR: KBr (4000–400 cm−1): 1612 υ(carbonyl); 3200–2860, 1725, 1703, 1583, 1560, 1444, 1368, 1310, 1275, 1212, 1173, 1096, 1036, 805. Mass Spec. (ESI positive ion): m/z: 827 [Co(C343)2(cyclam)]+.
Preparation of [Co(AQ2C)2(cyclam)]Cl·0.5H2O
Due to the light sensitivity of AQ2CH, all steps in the following procedure were carried out in the dark.AQ2CH (0.21 g, 0.84 mmol) was suspended in 30 ml of MeOH that had been dried over Na2SO4 and then triethylamine (151 μl, 0.84 mmol) was added. The mixture was then stirred for 5 min before the addition of [CoCl2(cyclam)]Cl (0.10 g, 0.27 mmol) as a solid and refluxed for 48 h. The crude solid was filtered off and recrystallised in DMF/H2O, the resulting solids were washed with MeOH (20 ml) and diethyl ether (10 ml) to give the product as a pale pink solid. (yield: 0.14 g, 0.18 mmol, 60.9%). Anal. Calc. for [Co(C15H7O4)2(C10H24N4)]Cl·0.5H20: C 59.60; H 4.88; N 6.95. Found C 59.71, H 5.82, N 6.41. IR: KBr (4000–400 cm−1): 3535, 3357, 3231, 2966, 2874, 1674, 1604, 1556, 1484, 1400, 1334, 1292, 1147, 1091, 937, 802, 790, 703. Mass Spec (ESI positive ion): m/z: 761 [Co(AQ2C)2(cyclam)]+.
Reduction in situ
Emission scans were run between 445 and 700 nm, with an excitation wavelength of 425 nm, with a scan rate of 100 nm min−1 and a 5 nm slit width at regular time intervals for up to three days on 50 μM solutions of [Co(AQ2C)2(cyclam)]Cl in DMA, following the addition of 10 molar equivalents of sodium ascorbate. These measurements were then repeated on solutions that had been deoxygenated by nitrogen gas for 15 min.Electrochemical studies
Cyclic voltammetry was conducted on a BAS 100B/W Electrochemical Analyser at a scan rate of 100 mV s−1, using a glassy carbon working electrode, a platinum auxiliary electrode, and a silver wire as a quasi-reference electrode. The experiments were performed at room temperature and were iR compensated. Samples were prepared as 1 mM solutions in DMF with 0.1 M tetrapropylammonium perchlorate and were degassed with argon for 10 min prior to measurement. The potentials were measured using ferrocene as an internal standard ([Fe(η5–C5H5)2]0/+ = +0.72 V vs.NHE in DMF34). E° is defined as the reduction potential, calculated by taking the mid-range potential of the fc/fc+ couple as 0 mV and determining the mid-range potential of the Co(III/II) couple.Construction of the 4xHRE-EosFP construct
The CMV promoter element was removed from the FLAG® tagged tetrameric wild-type EosFP (MoBiTec GmbH) by restriction digest with BglII/KpnI (Promega) and the annealed and phosphorylated oligos of the miniCMV promoter sequence (bp −51 to +77 of the CMV promoter)46 were inserted. This construct was then digested with ZraI (New England Biolabs) to allow insertion of the annealed, phosphorylated and ligated HRE element oligonucleotides.38 Confirmation of HRE copy number was confirmed by digestion with KpnI/SspI/XbaI and through DNA sequencing by the Australian Genome Research Facility (Westmead, Australia).Oligonucleotide sequences
All oligonucleotides were synthesized and PAGE purified by IDT Technologies (Coralville, IA, USA). The mini-CMV element was synthesized in two fragments and subsequently ligated after annealing and phosphorylation.HRE element forward: 5′gatcgccaca gtgcatacgt gggctccaac aggtcctctt ggatcgccac agtgcatacg tgggctccaa caggtcctct ta 3′. HRE element reverse: 5′gatctaagag gacctgttgg agcccacgta tgcactgtgg cgatccaaga ggacctgttg gagcccacgt atgcactgtg gcgatc 3′. Mini-CMV-1 forward: 5′gatctaggcg tgtacggtgg gaggcctata taagcagagc tcgtttagtg aaccgtcaga tcgcctg 3′. Mini-CMV-1 reverse: 5′ tctccaggcg atctgacggt tcactaaacg agctctgctt atataggcct cccaccgtac acgccta 3′. Mini-CMV-2 forward: 5′ gagacgccat ccacgctgtt ttgacctcca tagaagacac cgggaccgat ccagcctccg cggcccggta c 3′. Mini-CMV-2 reverse: 5′ cgggccgcgg aggctggatc ggtcccggtg tcttctatgg aggtcaaaac agcgtggatg gcg 3′.
Cell lines
DLD-1 human colon carcinoma cells were obtained from ATCC and used within 6 months of resuscitation. Cells were maintained in Advanced DMEM (Invitrogen) and supplemented with 2% FBS and 2 mM Glutamine in a humidified environment at 37 °C and 5% CO2. Cells were transfected using SuperFect Transfection Reagent (Qiagen) according to the manufacturer's instructions. Transfected cells were selected with 1 mg ml−1 of G418 (Invitrogen) and maintained in culture with 250 μg ml−1 of G418.IC50 determination
1 × 105cells ml−1 were seeded into each well of a 96-well plate and incubated under standard culturing conditions. [CoCl2(cyclam)]Cl was prepared as a 5 mM solution in deionised water. AQ2CH and [Co(AQ2C)2(cyclam)]Cl were prepared as 20 mM and 2 mM solutions in DMSO respectively. C343H and [Co(C343)2(cyclam)]Cl were prepared as a 2 mM solution in 60% (v/v) acetone immediately prior to the assay. All solutions were then filtered through a 25 mm Millex Syringe Filter (Millipore). Non-aqueous solvent concentrations were kept below 10% and these concentrations were verified as non-toxic. The dose volume of the cobalt complex solutions was kept below 40 μl and diluted in culture medium to be added to triplicate wells, spanning a 4-log range of final concentrations. The cells were then incubated for 72 h before 1 mM MTT was added to each well and further incubated for 4 h. The media was removed and 150 μl of DMSO was added to each well. The absorbance of each well was then measured at 600 nm after shaking for 1 min in a Victor3V microplate reader (Perkin Elmer). IC50 values were determined as the drug concentration that reduced the absorbance to 50% of that in untreated control wells.Drug treatments
AQ2C and [Co(AQ2C)2(cyclam)]Cl were freshly prepared in DMSO with C343H and [Co(C343)2(cyclam)]Cl freshly prepared in acetone/water (2:1) before treatment. Cell monolayers on glass coverslips were treated with 10 μM of compound for 1 h. The spheroids were treated with 10 μM of compound and incubated for 24 h.
Spheroid culture
Spheroids were formed by plating 100 μl of a 1.5 × 105cells ml−1 single cell suspension onto agarose coated 96 well imaging plates (BD Biosciences) and they were allowed to aggregate for 72 h resulting in the formation of a single spheroid per well.15 Spheroids were then dosed with 10 μM compound on day 4.Image acquisition
Confocal images were collected on the Olympus FV1000 inverted microscope using an Olympus UPLAPO 10x/0.40 air objective lens, UPLAPO 40x/1.00 oil objective lens and PLAPO 60x/1.40 water objective lens. A scan rate of 4.0 μs pixel−1, Kalman averaging and sequential collection of the green then blue channels were applied for all images. Excitation and emission ranges for the 405 nm diode, 488 nm argon and 559 nm diode lasers respectively were as follows: Ex 405 nm: Em 425–475 nm, Ex 488 nm: Em 500–550 nm, Ex 559 nm: Em 580–630 nm.Acknowledgements
The authors acknowledge funding support from the Australian Research Council (DP0773953 and DP110100461). The authors would like to thank Dr Renee Whan for technical assistance and acknowledge scientific and technical input and support from the Australian Microscopy & Microanalysis Research Facility (AMMRF) node at the University of Sydney.Notes and references
- J. M. Brown, Method Enzymol, 2007, 435, 297–321 CrossRef CAS
.
- P. Vaupel, F. Kallinowski and P. Okunieff, Cancer Res., 1989, 49, 6449–6465 CAS
- J. M. Brown and A. J. Giaccia, Cancer Res., 1998, 58, 1408–1416 CAS
- D. A. Tennant, R. V. Duran and E. Gottlieb, Nat. Rev. Cancer, 2010, 10, 267–277 CrossRef CAS
- K. DeClerck and R. C. Elble, Front. Biosci., 2010, 15, 213–225 CrossRef CAS
- M. Milani and A. L. Harris, Eur. J. Cancer, 2008, 44, 2766–2773 CrossRef CAS
- P. Subarsky and R. P. Hill, Clin. Exp. Metastasis, 2003, 20, 237–250 CrossRef CAS
- N. S. Kenneth and S. Rocha, Biochem. J., 2008, 414, 19–29 CrossRef CAS
- C. Brahimi-Horn and J. Pouysségur, B Cancer, 2006, 93, 73–80 Search PubMed
- I. F. Tannock, C. M. Lee, J. K. Tunggal, D. S. Cowan and M. J. Egorin, Clin Cancer Res, 2002, 8, 878–884 CAS
- P. Bischoff, A. Altmeyer and F. Dumont, Expert Opin. Ther. Pat., 2009, 19, 643–662 CrossRef CAS
- S. Koch, F. Mayer, F. Honecker, M. Schittenhelm and C. Bokemeyer, Br. J. Cancer, 2003, 89, 2133–2139 CrossRef CAS
- A. I. Minchinton and I. F. Tannock, Nat. Rev. Cancer, 2006, 6, 583 CrossRef CAS
- O. Tredan, C. M. Galmarini, K. Patel and I. F. Tannock, J. Natl. Cancer Inst., 2007, 99, 1441–1454 CrossRef CAS
- N. S. Bryce, J. Z. Zhang, R. M. Whan, N. Yamamoto and T. W. Hambley, Chem. Commun., 2009, 2673–2675 RSC
- W. R. Wilson, J. W. Moselen, S. Cliffe, W. A. Denny and D. C. Ware, Int. J. Radiat. Oncol., Biol., Phys., 1994, 29, 323–327 CrossRef CAS
- D. C. Ware, B. D. Palmer, W. R. Wilson and W. A. Denny, J. Med. Chem., 1993, 36, 1839–1846 CrossRef CAS
- J. B. J. Milbank, R. J. Stevenson, D. C. Ware, J. Y. C. Chang, M. Tercel, G. O. Ahn, W. R. Wilson and W. A. Denny, J. Med. Chem., 2009, 52, 6822–6834 CrossRef CAS
- W. A. Denny, Future Oncol., 2010, 6, 419–428 CrossRef CAS
-
S. J. Lippard and J. M. Berg, Principles of Bioinorganic Chemistry, University Science Books, 1994 Search PubMed
- R. F. Anderson, W. A. Denny, D. C. Ware and W. R. Wilson, Br J Cancer Suppl, 1996, 27, S48–51 CAS
- P. R. Craig, P. J. Brothers, G. R. Clark, W. R. Wilson, W. A. Denny and D. C. Ware, Dalton Trans., 2004, 611–618 RSC
- T. Salthammer and H. Dreeskamp, J. Photochem. Photobiol., A, 1990, 55, 53–62 CrossRef CAS
- J. A. Kemlo and T. M. Shepherd, Chem. Phys. Lett., 1977, 47, 158–162 CrossRef CAS
-
S. Danos, The University of Sydney, 2005
- M. D. Hall, T. W. Failes, N. Yamamoto and T. W. Hambley, Dalton Trans., 2007, 3983–3990 RSC
- N. Yamamoto, S. Danos, P. D. Bonnitcha, T. W. Failes, E. J. New and T. W. Hambley, JBIC, J. Biol. Inorg. Chem., 2008, 13, 861–871 CrossRef CAS
- N. J. Wheate, C. R. Brodie, J. G. Collins, S. Kemp and J. R. Aldrich-Wright, Mini-Rev. Med. Chem., 2007, 7, 627–648 CrossRef CAS
- S. O. Mueller, W. K. Lutz and H. Stopper, Mutat Res, 1998, 414, 125–129 CAS
- A. K. Shinhababu and D. R. Thakker, Adv. Drug Delivery Rev., 1996, 19, 241–273 CrossRef
- S. Manazanaro, M. J. Vincent, M. J. Martin, N. Salvador-Tormo, J. M. Pérez, M. del Mar Blanco, C. Avendaño, J. C. Menéndez and J.Á. de la Fuente, Bioorg. Med. Chem., 2004, 12, 6505–6515 CrossRef
- E. Reisner, V. B. Arion, B. K. Keppler and A. J. L. Pombeiro, Inorg. Chim. Acta, 2008, 361, 1569–1583 CrossRef CAS
- O. Reza, J. Raoof and M. Ebrahimi, Iran J Chem Chem Eng, 2001, 20, 75–81 Search PubMed
- W. C. Barrette, H. W. Johnson and D. T. Sawyer, Anal. Chem., 1984, 56, 1890–1920 CrossRef CAS
- M. Xia, K. Bi, R. Huang, M.-H. Cho, S. Sakamuru, S. Miller, H. Li, Y. Sun, J. Printen, C. Austin and J. Inglese, Mol. Cancer, 2009, 8, 117–129 CrossRef
- K. Shield, M. L. Ackland, N. Ahmed and G. E. Rice, Gynecol. Oncol., 2009, 113, 143–148 CrossRef
- J. M. Padrón, C. L. van der Wilt, K. Smid, E. Smitskamp-Wilms, H. H. J. Backus, P. E. Pizao, G. Giaccone and G. J. Peters, Crit. Rev. Oncol. Hematol., 2000, 36, 141–157 CrossRef
- D. E. Post and E. G. Van Meir, Gene Ther., 2001, 8, 1801–1807 CrossRef CAS
- M. Chen, M. Gupta and K. Cheung, Biomed. Microdevices, 2010, 12, 647–654 CrossRef CAS
- J. Friedrich, C. Seidel, R. Ebner and L. A. Kunz-Schughart, Nat. Protoc., 2009, 4, 309–324 CrossRef CAS
- R. Herrmann, W. Fayad, S. Schwarz, M. Berndtsson and S. Linder, J. Biomol. Screening, 2008, 13, 1–8 CrossRef CAS
- P. Iglesias and J. A. Costoya, Biosens. Bioelectron., 2009, 24, 3126–3130 CrossRef CAS
- I. Serganova, M. Doubrovin, J. Vider, V. Ponomarev, S. Soghomonyan, T. Beresten, L. Ageyeva, A. Serganov, S. Cai, J. Balatoni, R. Blasberg and J. Gelovani, Cancer Res., 2004, 64, 6101–6108 CrossRef CAS
- J. Wiedenmann, S. Ivanchenko, F. Oswald, F. Schmitt, C. Rocker, A. Salih, K. D. Spindler and G. U. Nienhaus, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15905–15910 CrossRef CAS
- B. Bosnich, M. L. Tobe and G. A. Webb, Inorg. Chem., 1965, 4, 1109–1112 CrossRef CAS
- M. Boshart, F. Weber, G. Jahn, K. Dorsch-Hasler, B. Fleckenstein and W. Schaffner, Cell, 1985, 41, 521–530 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: experimental procedures and 1 supplementary figure. See DOI: 10.1039/c1sc00337b |
| This journal is © The Royal Society of Chemistry 2011 |