A practical, convergent route to the key precursor to the tetracycline antibiotics†
David A.
Kummer‡
,
Derun
Li‡
,
Amelie
Dion
and
Andrew G.
Myers
*
Department of Chemistry and Chemical Biology, Harvard University, Cambridge, Massachusetts 02138, USA. E-mail: myers@chemistry.harvard.edu; Fax: +1-617-495-4976; Tel: +1-617-495-5679
First published on 7th July 2011
Abstract
Here we describe a 5-step sequence to prepare the AB enone 1, the key precursor to fully synthetic tetracyclines, that begins with a diastereoselective Michael–Claisen coupling of two simple starting materials, a cyclohexenone (compound 2 or, in a refinement, a substituted variant, vide infra) and the isoxazole ester 3. This advance defines an 8-step linear sequence to 6-deoxytetracycline antibiotics from three components of similar complexity (cyclohexenone 2, isoxazole ester 3, and structurally diverse D-ring precursors) in which sequential diastereoselective Michael–Claisen cyclization reactions form the A- and C-rings, respectively, of the linearly fused ABCD tetracycline skeleton. In addition to providing a readily scalable, practical route to fully synthetic tetracyclines of broad structural diversity, the sequence reported comprises a series of non-obvious stereoselective transformations, including a novel means for C12a hydroxylation.
Introduction
Previously we showed that the AB enone 1 serves as a highly versatile intermediate for the construction of diverse tetracycline antibiotics.1,2 This key precursor can be transformed into fully deprotected synthetic 6-deoxytetracyclines in a sequence of just 3 steps (illustrated by the synthesis of 6-deoxytetracycline, Scheme 1). A focal point in the production of novel tetracycline antibiotics in amounts necessary for drug discovery and manufacturing efforts then has been the development of a practical and efficient route to the AB enone 1. To date, we have reported two different enantioselective routes to this substance. The first proceeded by an 11-step linear sequence from benzoic acid as starting material (10% yield) and provided gram-quantities of the crystalline AB enone 1, but was challenging to scale in larger amounts.1 A second, more convergent process proceeded in 5-steps (29% yield) from the coupling of two precursors of similar complexity [3-methoxyfurfural (4) and the isoxazole 5, Scheme 2] and has proven to be more amenable to large-scale synthesis of 1.3 Detailed here is an entirely new strategy for the convergent assembly of the AB enone 1, employing different coupling components, derived by retrosynthetic disconnection of 1 at positions distinct from those targeted in earlier approaches. The route features a highly diastereoselective Michael–Claisen cyclization reaction upon an existing B ring precursor, forming in one step two carbon–carbon bonds and the A ring. Stereocontrolled transformations of the (AB) cyclization product were then developed, relaying the stereochemistry of position C4 (initially non-homologous with natural tetracyclines, tetracycline numbering) to position C12a, this by a later-stage hydroxylation reaction, and then back to C4, correcting the stereochemistry of this position. The present process can be readily scaled and offers many favorable features in this regard, such as the crystallinity of intermediates at many stages of the process (allowing for purification without resort to chromatography) as well as an efficient recycling protocol (vide infra).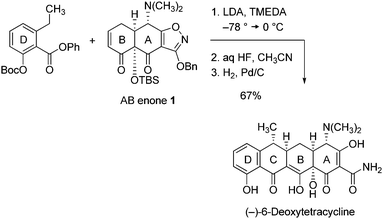 | ||
| Scheme 1 Construction of (−)-6-deoxytetracycline by Michael–Claisen cyclization.1,2 | ||
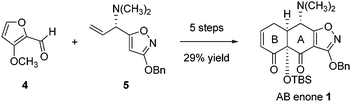
Retrosynthetic analysis
In light of the efficiency and generality of the late-stage Michael–Claisen cyclization reaction that was used to construct the C ring of tetracyclines (Scheme 1), we were led to consider whether this powerful transformation might also be applied to assemble the AB enone 1 and specifically its A ring. Two obstacles prevented direct application of a Michael–Claisen disconnection within target 1. The first was the tert-butyldimethylsilyloxy group at position C12a, which it seemed would have to be cleared to provide a viable retron (Scheme 3), leading us to evaluate the feasibility of C12a oxygenation by hydroxylation of a substrate such as 6. That C12a hydroxylation might not proceed with proper stereochemistry was an issue of initial concern, but one that did not materialize in practice, where we observed that a stereoisomer of compound 6 (the C4-epimer, structure 13, Scheme 7, vide infra) was formed in the sequence we present below. This proved fortuitous, for highly stereoselective hydroxylation was achieved with this substrate, in the desired sense. Second, beyond the issue of C12a hydroxylation was the problem of identifying a suitable equivalent of 2,5-cyclohexadien-1-one, which was clearly not a viable B-ring precursor in light of its propensity to aromatize (Scheme 3).4 Contributions from the Ogasawara–Takano laboratories over many years have defined a practical and versatile equivalent of 2,5-cyclohexadien-1-one in the form of its cyclopentadiene-Diels–Alder adduct 2. Through their research this compound is now readily available in both enantiomeric forms.5–7 Ogasawara, Takano, and co-workers, as well as other research groups, have shown that the versatile building block 2 can be transformed into an extraordinary variety of structures in just a few steps, one of these being an obligate high-temperature retro-Diels–Alder reaction that expels cyclopentadiene.8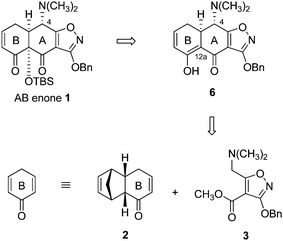 | ||
| Scheme 3 A Michael–Claisen cyclization approach to A-ring construction. | ||
By the analysis above, the specific transformation we were led to explore initially was the Michael–Claisen condensation reaction of the Ogasawara–Takano cyclohexenone 2 and methyl 3-benzyloxy-5-(dimethylaminomethyl)isoxazole-4-carboxylate (3). We refer to the latter compound as the Stork–Hagedorn isoxazole, recognizing the first reported use of the 3-benzyloxyisoxazole function as a protective group for the A-ring of tetracyclines, an innovation critical to our own work.9 The Stork laboratory subsequently described a remarkably efficient synthesis of C12a-deoxytetracycline from this same starting material using stepwise Michael and Claisen cyclization reactions to form the A-ring, with a different sequencing than that reported here.10 In the present application, neither the feasibility nor the likely stereochemical outcome of the proposed single-step condensation (2 + 3) were known at the outset of our work. In the following section, we detail a practical and short sequence for the synthesis of the AB enone 1. We also describe further innovations in the route, including the development of a modified Ogasawara–Takano enone (containing an 11-dimethylphenylsilyl substituent, structure 25, Scheme 9, vide infra), which has permitted large-scale synthesis of 1 in pure form without resorting to column chromatography.
Results and discussion
The Ogasawara–Takano cyclohexenone 2 was synthesized in three steps by simple modifications of procedures originally reported for the preparation of this substance (Scheme 4).5 Thus, reduction of the cyclopentadiene-benzoquinone Diels–Alder adduct 711 with sodium borohydride (1.2 equiv) in the presence of cerium chloride (2 equiv) in methanol provided the mesodiol 8 in 97% yield.12,13 Enantioselective acylation of 8 was best accomplished using an immobilized Amano lipase (Pseudomonas sp.) and isopropenyl acetate (3 equiv) as the acetylating agent in triethylamine as solvent. The use of triethylamine alone as the solvent and isopropenyl acetate as the acetylating agent represent departures from the original protocols.5,7 By using the combination of lipase, acetylating agent, and solvent specified we observed complete transformation of 8 within 22 h, and obtained the monoacetate 9 free of any bis-acylated material, a by-product we observed to form in small amounts (1–5% yield) when vinyl acetate was used as the acetylating agent.5,7 The immobilized lipase could be recovered by filtration of the product mixture through a sintered-glass funnel, rinsing the filter cake with ethyl acetate, and then drying the rinsed lipase in vacuo (0.1 mmHg) at 23 °C. The recovered, dried lipase could be recycled over at least two iterations without diminution in the yield or enantioselectivity of the monoacetate product (9). As reported by Ogasawara and Takano, exposure of monoacetate 9 to dichlorobis(triphenylphosphine)palladium(II) (1 mol %) in the presence of ammonium formate (1.25 equiv) in refluxing acetonitrile afforded the cyclohexenone 2 in 79% yield after purification by chromatography on silica gel (>99% ee, chiral GC analysis).5,14 This practical 3-step sequence to the cyclohexenone 2 did require final purification by column chromatography in order to achieve optimal results in subsequent transformations, a purification step not practiced when using a silyl-substituted variant of cyclohexenone 2, discussed below. The cyclohexenone 2 was liquid at 23 °C and solidified at −20 °C. Typically, 30–35-g batches of cyclohexenone 2 were prepared.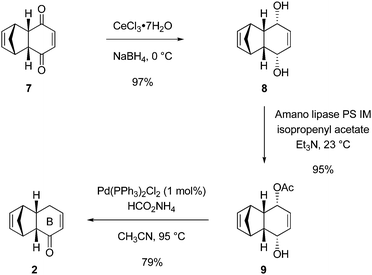 | ||
| Scheme 4 Synthesis of cyclohexenone 2. | ||
The Stork-Hagedorn isoxazole (3)9,10 was readily prepared in large amounts by the sequence outlined in Scheme 5, beginning with the known substance methyl 3-hydroxyisoxazole-5-carboxylate (10) (see Supporting Information for details†).15,16
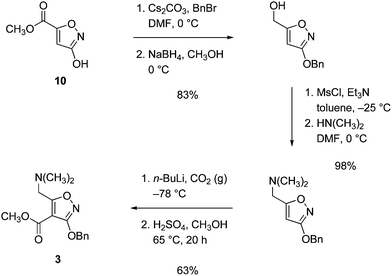 | ||
| Scheme 5 Synthesis of isoxazole ester 3. | ||
To form the A-ring of the AB enone 1, the isoxazole ester 3 (130 g, 1.05 equiv) was deprotonated with sodium hexamethyldisilazide (1.05 equiv) in tetrahydrofuran (−78 °C for 20 min, −20 °C for 1 h, then re-cooling to −78 °C). The resulting thick slurry was stirred mechanically at −78 °C while a solution of the cyclohexenone 2 (68.0 g, 1 equiv) was added over the course of 45 min, leading to formation of the Michael addition product (thin-layer chromatographic analysis, not characterized, Scheme 6). After 30 min, a solution of potassium hexamethyldisilazide (1.05 equiv) in toluene was added over 70 min so as to ensure that the Claisen product was deprotonated rapidly as it was formed during the subsequent warming step. Failing this, the cyclization reaction was observed not to proceed to completion, presumably due to quenching of the enolate intermediate (11) by proton transfer from the 1,3-diketone product or methanol. Claisen cyclization proceeded upon warming to −20 °C and was complete within 6 h. After an aqueous workup and purification by flash column chromatography on silica gel the Michael–Claisen cyclization product 12 was obtained as a pale yellow foam (143 g, 80%). 1H NMR analysis (CDCl3) revealed that the product was entirely enolic and, within the limits of detection, a single diastereomer.17
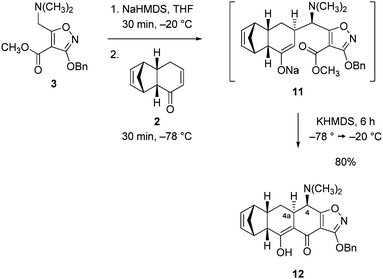 | ||
| Scheme 6 Michael–Claisen cyclization reaction of components 2 and 3. | ||
Analysis of proton–proton coupling constants strongly supported the stereochemical assignments depicted in structure 12; these were confirmed by later transformations and, ultimately, X-ray crystallographic analysis (vide infra). Interestingly, when we evaluated the condensation of the cyclohexenone 2 with the corresponding enolate derived from the isoxazole phenyl ester as the nucleophile (in lieu of the enolate derived from the methyl ester) under conditions otherwise identical to those described above, the rates of the Michael and Claisen reactions were observed to be similar, and the yield of the Michael–Claisen cyclization product was lower (44%), presumably a consequence of competing proton transfer. We believe that the higher yield of the Michael–Claisen condensation of the enolate derived from the methyl ester 3 with (essentially an equimolar amount of) the cyclohexenone coupling component 2 is in large measure due to the fact that the Claisen cyclization reaction is rate determining with these substrates.
Michael addition proceeds by addition of the sodium enolate to the sterically more accessible face of cyclohexenone 2 as expected based on precedent8 and, strikingly, with complete control of the relative stereochemistry of the C4 and C4a centers. This provides an additional instance of high diastereocontrol in the creation of adjacent stereogenic sp3-centers by Michael–Claisen cycloaddition (another being the formation of the C-ring, exemplified in Scheme 1), suggesting that this powerful ring-forming reaction may be more broadly applicable as a method for the stereocontrolled construction of 6-membered rings. Interestingly, comparison of the cycloaddition reactions of Scheme 1 and Scheme 6 reveals that they are non-homologous stereochemically, if the methyl and dimethylamino substituents of the respective enolate intermediates are given equal prioritization. In both cases nucleophilic addition to the β-position of each cyclohexenone is believed to occur along a pseudoaxial trajectory, but this pathway is believed to be sterically much more restricted in the case of the AB enone 1 relative to the cyclohexenone 2 (the C4 position of enone 1 presents a pseudo 1,3-diaxial interaction during Michael addition). The tert-butyldimethylsilyloxy substituent at position C12a is believed to block nucleophilic addition to the α-face of enone 1, much as the cyclopentadienyl residue blocks addition to the α-face of cyclohexenone 2.8 The enolates are of course also quite different, having different counterions, as well as substituents with potentially quite different propensities for metal (counterion) binding, and this, too, may play a role in the differing stereochemical outcomes of A- and C-ring formation.
Although the stereochemistry of position C4 within the Michael–Claisen cyclization product 12 did not correspond with that of naturally occurring tetracyclines, we imagined that this product might serve as a viable precursor to the AB enone 1 nevertheless, provided that epimerization at position C4 could be achieved subsequently. The most viable sequence appeared to be: (1) expulsion of cyclopentadiene from 12 by retro-Diels–Alder reaction [hydroxylation of 12 (or its C4 epimer), if it could be achieved at all, would almost certainly not proceed with correct stereochemistry at C12a due to steric blocking by the cyclopentenyl group] (2) hydroxylation of the retro-Diels–Alder product 13, and (3) C4-epimerization. As discussed in the introduction, having obtained the “incorrect” stereochemistry at position C4 during the Michael–Claisen cycloaddition reaction was likely advantageous, as it was unclear that C12a-hydroxylation could have been achieved in the desired sense at any stage had C4 been set with the “correct” stereochemistry.
In accord with literature precedent,18 retro-Diels–Alder fragmentation of the Michael–Claisen cyclization product 12 proceeded rapidly only at temperatures above 200 °C. As a complication, the cyclohexenone produced (13, Scheme 7) was observed to undergo partial decomposition under the conditions of thermolysis and this appeared to be exacerbated at higher concentrations of substrate (12).19 The optimized protocol for thermolysis then involved a short contact time at elevated temperature and a relatively dilute substrate solution. Specifically, a solution of substrate 12 (143 g, 0.02 M) and dimethyl maleate (213 mL, 5 equiv, added as a trapping agent for cyclopentadiene) in diphenyl ether (14 L) was pumped continuously using a peristaltic pump through a stainless-steel tube submerged in an oil bath heated to 250 °C at a flow rate of ∼12 mL min−1 (Scheme 7, for details of the apparatus, including photographs, see Supporting Information†). Addition of hydrogen chloride to the cooled product solution led to precipitation of the product as its hydrochloride salt. This was collected, the corresponding free base was formed, and the latter (13) was purified by a combination of crystallization (31.0 g, yellow prisms, mp 103–105 °C, 1st crop) and flash column chromatography (36.0 g, yellow solid, mp 103–105 °C, 2nd crop). The total yield of pure retro-Diels–Alder product 13 was 55% (67.0 g). The crystals collected in the 1st crop were suitable for X-ray analysis. The solid-state structure we obtained, depicted in Fig. 1, provided confirmation of stereochemistry and a basis for optimism that C12a-hydroxylation of 13 (or more properly an anion derived from it) might proceed by selective attack of the oxidant on the α-face, for the β-face appeared to be largely blocked by the C4-dimethylamino group. The high-temperature, continuous-flow retro-Diels–Alder reaction we developed was not easily reproduced without meticulous attention to detail in setup of the specialized reaction apparatus and this, coupled with the large volume of high-boiling solvent required, compelled us to develop an alternative protocol (vide infra), but first we sought to establish that intermediate 13 could serve as a precursor to the AB enone 1.
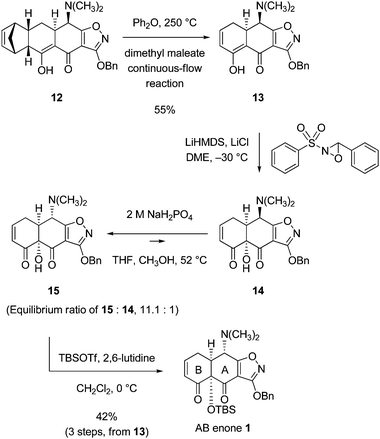 | ||
| Scheme 7 Synthesis of AB enone 1. | ||
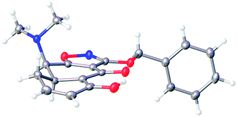 | ||
| Fig. 1 X-ray crystal structure of the retro-Diels–Alder product 13. | ||
An extensive literature describes with somewhat mixed success late-stage C12a-hydroxylation reactions in tetracycline synthesis, including each of the formative studies in the field. In the first fully synthetic route to any tetracycline antibiotic, (d,l-6-demethyl-6-deoxytetracycline, sancycline, 17), Woodward and co-workers reported that d,l-6-demethyl-6,12-dideoxytetracycline (16) was hydroxylated in the presence of dioxygen and cerous chloride in buffered methanol-DMF to provide synthetic sancycline in 6.5% yield (Scheme 8).20 In their synthesis of d,l-terramycin, the Muxfeldt laboratory described a more efficient C12a-hydroxylation reaction (18 → 19, 52% yield) using a different protocol, also employing dioxygen as the stoichiometric oxidant.21 A later publication from the Stork laboratories reported that conditions could not be found to achieve late-stage C12a hydroxylation of 12a-deoxytetracycline to produce tetracycline itself (20 → 21).10 We recognized that C12a-hydroxylation of the retro-Diels–Alder product 13 is different contextually than the examples in the references cited, for the vinylogous carbamic acid group of the tetracyclines is masked in the form of the Stork-Hagedorn isoxazole residue9 and, of course, the substrate we imagined for hydroxylation (13) lacks the extended conjugation within the C- and D-rings that is characteristic of the substrates in Scheme 8.
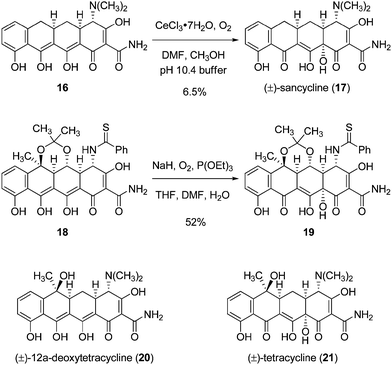 | ||
| Scheme 8 Late-stage C12a-hydroxylation reactions in tetracycline synthesis.20,21,10 | ||
In the present case, stereoselective C12a hydroxylation of substrate 13 was best achieved using as stoichiometric oxidant a reagent other than dioxygen. In the protocol we first employed for large-scale hydroxylation of 13 (a modified procedure is presented below) the Davis sulfonyloxaziridine22 (60.5 g, 1.32 equiv) was added portion-wise as a slurry in 1,2-dimethoxyethane (DME) over 1 h to a mechanically stirred, cold (−30 °C) slurry of an enolate obtained by deprotonation of 13 (67.0 g, 1 equiv) in the presence of anhydrous lithium chloride (1.1 equiv) in DME at −30 °C using as base a freshly prepared solution of lithium hexamethyldisilazide (1.1 equiv) in DME. After an aqueous workup involving extraction of the product into water as its hydrochloride salt, then formation of the corresponding free base, concentration afforded the C12a-hydroxylation product 14 as a purple foam, estimated by 1H NMR analysis to be >95% a single diastereoisomer (48.0 g, 68% crude yield, an accurate yield is reported after the subsequent steps, C4-epimerization and hydroxyl protection).
It has long been known that position C4 of tetracyclines is subject to epimerization under various conditions. Scientists at Lederle laboratories reported in 1955 that tetracycline undergoes equilibration in 1 M aqueous sodium dihydrogen phosphate-methanol (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 25 °C, 24 h) to afford a 1.5:1 mixture of natural (4S) and nonnatural (4R) stereoisomers, respectively.23 Noseworthy,24 and later Woodward in collaboration with Pfizer scientists, showed that under somewhat different conditions (CaCl2, water-butanol containing ethanolamine, pH 8.5, reflux, 3.5 h) a mixture of C4-epimers of sancycline is isomerized to afford predominately the natural (4S)-stereoisomer.20 These examples are somewhat distantly removed from the substrate of present interest, the (4R)-C12a hydroxylation product 14. In the event, when a solution of the latter was heated at 52 °C for 15 h in a mixed solvent containing tetrahydrofuran (2 parts), methanol (2 parts), and 2 M aqueous sodium dihydrogen phosphate solution (3 parts) almost complete isomerization to the desired 4S-epimeric product (15) occurred. The ratio of desired to undesired epimers was 11.1 to 1. The diastereomers could be separated by column chromatography; resubjection of the pure desired (4S)-isomer to the conditions of epimerization gave rise to the same 11.1:1 ratio of epimers obtained from the (4R)-isomer, which establishes that this is the equilibrium ratio under the conditions defined (later, we show that the equilibrium ratio is improved under slightly modified conditions). In practice, the diastereomeric mixture was not separated. Rather, the mixture was silylated directly (TBSOTf, 2,6-lutidine, CH2Cl2, 0 °C) to provide the AB enone 1. The undesired (4R)-epimer 14 does not undergo silylation under these reaction conditions and is easily separated from the AB enone 1. The three steps (13 → 14 → 15 → 1) were conducted in sequence with final chromatographic purification on silica gel; the yield for the three steps was 42%. In the largest single-batch implementation of the 5-step sequence outlined (beginning with the coupling of the cyclohexenone 2 and the isoxazole ester 3) we prepared 39.1 g of the AB enone 1 in 18% yield overall. Three chromatographic purification steps were necessary.
:1, 25 °C, 24 h) to afford a 1.5:1 mixture of natural (4S) and nonnatural (4R) stereoisomers, respectively.23 Noseworthy,24 and later Woodward in collaboration with Pfizer scientists, showed that under somewhat different conditions (CaCl2, water-butanol containing ethanolamine, pH 8.5, reflux, 3.5 h) a mixture of C4-epimers of sancycline is isomerized to afford predominately the natural (4S)-stereoisomer.20 These examples are somewhat distantly removed from the substrate of present interest, the (4R)-C12a hydroxylation product 14. In the event, when a solution of the latter was heated at 52 °C for 15 h in a mixed solvent containing tetrahydrofuran (2 parts), methanol (2 parts), and 2 M aqueous sodium dihydrogen phosphate solution (3 parts) almost complete isomerization to the desired 4S-epimeric product (15) occurred. The ratio of desired to undesired epimers was 11.1 to 1. The diastereomers could be separated by column chromatography; resubjection of the pure desired (4S)-isomer to the conditions of epimerization gave rise to the same 11.1:1 ratio of epimers obtained from the (4R)-isomer, which establishes that this is the equilibrium ratio under the conditions defined (later, we show that the equilibrium ratio is improved under slightly modified conditions). In practice, the diastereomeric mixture was not separated. Rather, the mixture was silylated directly (TBSOTf, 2,6-lutidine, CH2Cl2, 0 °C) to provide the AB enone 1. The undesired (4R)-epimer 14 does not undergo silylation under these reaction conditions and is easily separated from the AB enone 1. The three steps (13 → 14 → 15 → 1) were conducted in sequence with final chromatographic purification on silica gel; the yield for the three steps was 42%. In the largest single-batch implementation of the 5-step sequence outlined (beginning with the coupling of the cyclohexenone 2 and the isoxazole ester 3) we prepared 39.1 g of the AB enone 1 in 18% yield overall. Three chromatographic purification steps were necessary.
For the reasons outlined above we sought to modify the 5-step sequence in order to avoid the high-temperature retro-Diels–Alder reaction. A number of different strategies have been described to modify the diene-derived component of norbornenyl Diels–Alder adducts so as to facilitate their thermal fragmentation.18 Modified dienes include 1,1-dimethylfulvene in the early research of Alder and co-workers25 and later by Ichihara and co-workers,265-trimethylsilylcyclopentadiene in the research of Magnus and co-workers and applied by them in the synthesis of aspidosperma alkaloids,27pentamethylcyclopentadiene in the research of Grieco and co-workers and applied by them in the synthesis prostaglandins,28 and spiro[2.4]hepta-4,6-diene in the research of Kotha and Sunoj.29 Of these, the use of 5-trimethylsilylcyclopentadiene reported by Magnus and co-workers27 was particularly appealing because of its simplicity and the possibility that the 5-silylcyclopentadiene might be recycled (5-trimethylsilylcyclopentadiene is known to be greatly favored over the 1- and 2-trimethylsilylcyclopentadiene isomers at equilibrium).30
In the present work both 5-trimethylsilylcyclopentadiene and 5-dimethylphenylsilylcyclopentadiene were examined as substrates. We observed that intermediates containing the dimethylphenylsilyl substituent had a greater propensity to be crystalline, and so our focus turned to this specific substitution. 5-Dimethylphenylsilylcyclopentadiene (22a) was prepared using the protocol of Landais and Parra-Rapado for guidance.31Lithium cyclopentadienide, formed from freshly cracked cyclopentadiene (58.1 g, 1 equiv) and n-butyllithium (1 equiv), was trapped at −78 °C with dimethylphenylchlorosilane (150 g, 1 equiv) to provide (after an aqueous workup that involved only brief warming to 23 °C) in quantitative yield (175 g) a kinetic distribution of dimethylphenylsilylcyclopentadienes that contained approximately 92% of the 5-dimethylphenylsilylcyclopentadiene (22a) isomer (Scheme 9). The mixture of dienes was liquid at 23 °C and solid at −80 °C. The diene mixture was stored at −80 °C to prevent isomerization.
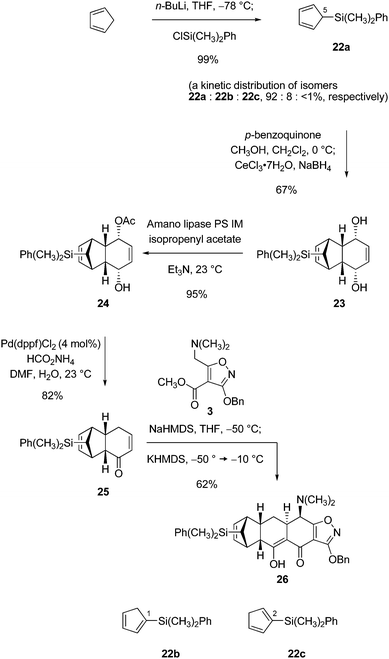 | ||
| Scheme 9 Introduction of the dimethylphenylsilyl group. | ||
Combination of the diene mixture (124 g, 92% 22a, 539 mmol 22a, 1 equiv 22a) and benzoquinone (63.3 g, 1.03 equiv) in a 2:1 mixture of methanol and dichloromethane at 0 °C led to complete cycloaddition within 12 h to form the expected 11-dimethylphenylsilyl endo enedione adduct (not depicted). The product was not isolated at this stage; rather, cerium trichloride heptahydrate (109 g, 0.51 equiv) and sodium borohydride (22.2 g, 1.03 equiv) were added in sequence, affording the mesodiol 23, which was readily isolated in pure form by recrystallization from dichloromethane–hexanes (129 g, 67% yield from the total mass of 124 g, 73% yield based on the amount of 22a present, mp 98–100 °C). Enzymatic desymmetrization of the mesodiol 23 (150 g) was achieved using the same immobilized lipase as above (Amano, Pseudomonas sp., 150 g) and isopropenyl acetate (3.5 equiv) as the acetylating agent in triethylamine as solvent to form monoacetate 24, which was isolated as a white solid (159 g, 95% yield, mp 68–72 °C) after decolorization with charcoal (Darco G-60).
Palladium-catalyzed rearrangement of the monoacetate product (24) was optimally conducted by substantial modification of the protocol of Ogasawara and Takano.5,14 In exploratory experiments, where we evaluated the suitability of the conditions originally developed by Ogasawara and Takano to transform the monoacetate 9 to the cyclohexenone 2 (Scheme 4 above) for the parallel transformation of the dimethylphenylsilyl-substituted monoacetate 24 to the cyclohexenone 25, we observed that phenol was formed as a substantial by-product and this appeared to be accelerated at elevated reaction temperatures (>30 °C). Presumably, this arose by a formal retro-Diels–Alder fragmentation of the cyclohexenone 25, although step-wise mechanisms (including palladium-mediated processes) can also be envisioned to transform 25 to phenol. By substantial modification of the catalyst and solvent we were able to effectively suppress the formation of phenol. Specifically, the use of [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II) (4 mol%) as catalyst in a 12:1 solvent mixture of dimethylformamide and water at 23 °C led to complete and clean transformation to the cyclohexenone 25 within 16 h. The reaction mixture was diluted with a 1:1 mixture of methyl tert-butyl ether and water and the resulting biphasic solution was stirred with charcoal (Darco G-60) at −10 °C for 1 h. The whole mixture was filtered through a pad of Celite, the filtrate was subjected to an aqueous workup, and the organic layer was concentrated, affording a brown solid. The cyclohexenone 25 was observed to decompose upon exposure to silica gel or basic alumina; in both cases phenol was formed.32 In lieu of purification by column chromatography, the crude solid product was dissolved in methyl tert-butyl ether and the resulting solution was stirred over a resin containing 2,4,6-trimercaptotriazine immobilized on a polystyrene solid support (MP-TMT, Biotage) at 23 °C for 16 h. Filtration through a sintered-glass funnel and then concentration of the filtrate afforded the cyclohexenone 25 as an orange solid (124 g, 82% crude yield, est. >90% purity by 1H NMR analysis).
An adaptation of the protocol developed for the Michael–Claisen cyclization of cyclohexenone 2 and isoxazole ester 3 (Scheme 6 above) was applied for the coupling of dimethylphenylsilyl-substituted cyclohexenone 25 and isoxazole ester 3. Thus, a solution of cyclohexenone 25 (124 g, 1 equiv) in tetrahydrofuran was added over the course of 40 min to a mechanically stirred slurry of the sodium enolate derived from the isoxazole ester 3 (134 g, 1.1 equiv) obtained in this case by deprotonation with sodium hexamethyldisilazide (1.15 equiv) in tetrahydrofuran at −50 °C. Formation of the Michael adduct appeared to be complete within 1 h at −50 °C (based on TLC analysis, not characterized), and, as before, potassium hexamethyldisilazide (1 equiv) was added to ensure that the Claisen product was deprotonated rapidly as it was formed. Claisen cyclization in the present transformation was initiated by warming to −20 °C over the course of 1 h and proceeded to completion within 2 h at −20 °C; the reaction solution was additionally held for 2 h at −10 °C. After an aqueous workup, addition of hydrogen chloride led to formation of a hydrochloride salt, which was recrystallized from dichloromethane–ethyl acetate (off-white solid, mp 158–160 °C). X-ray crystallographic analysis established that this intermediate was stereochemically homologous with the non-silylated intermediate 12 above (Fig. 2). The cycloadduct 26 was isolated as a white foam upon formation of its free base (146 g, 62% yield overall).
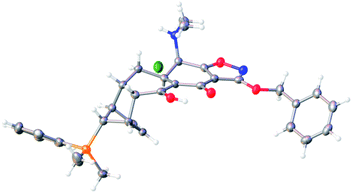 | ||
| Fig. 2 X-ray crystal structure of the hydrochloride salt of 26. | ||
In our preferred sequence for retro-Diels–Alder reaction, the Michael–Claisen cyclization product 26 (85.0 g) was heated in toluene at reflux for 10 h (Scheme 10), producing an equilibrium distribution of dimethylphenylsilylcyclopentadiene isomers (detailed below) and the desired retro-Diels–Alder product (13). The latter was obtained as yellow prisms by concentration of the reaction mixture and crystallization of the residue from ethyl acetate–hexanes (38.0 g, mp 103–105 °C, 1st crop). The isomeric dimethylphenylsilylcyclopentadienes were readily isolated by filtration of the mother liquors through a pad of silica gel, eluting with hexanes. Additional retro-Diels–Alder product 13 was obtained by further elution of the silica pad with ethyl acetate–hexanes, concentration of the eluent, and recrystallization of the solid product from ethyl acetate–hexanes to afford a second crop of crystals (9.40 g, mp 103–105 °C). The total amount of crystalline retro-Diels–Alder product 13 obtained was 47.4 g (87% yield).
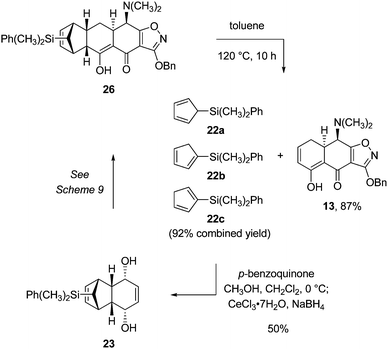 | ||
| Scheme 10 Retro-Diels–Alder reaction with dimethylphenylsilyl substitution. | ||
Concentration of the hexanes eluent containing the isomeric dimethylphenylsilylcyclopentadienes afforded 92% (28.5 g) of the theoretical mass, comprised of the following isomeric distribution (1H NMR analysis): 22a (75%), 22b (18%), 22c (7%). It was possible to separate the 5-dimethylphenylsilylcyclopentadiene isomer (22a) from the isomeric pair 22b and 22c by careful column chromatography. We separately subjected the samples enriched in the 5-dimethylphenylsilylcyclopentadiene isomer (22a) and the mixture of isomers 22b and 22c to conditions of reflux in toluene (10 h, bath temperature 120 °C) and in each case obtained the identical distribution of isomers (within experimental error) listed above, which therefore must represent the equilibrium distribution at ∼120 °C (see Supporting Information†). This is clearly advantageous in the present context, for it allows for recycling of the preponderance of the dimethylphenylsilylcyclopentadienyl retro-Diels–Alder product. In practice, combination of the unpurified dimethylphenylsilylcyclopentadiene mixture as obtained (28.5 g, 75% 22a, ∼107 mmol 22a, 1 equiv 22a) with benzoquinone (1.26 equiv) in methanol-dichloromethane at 0 °C formed the 11-dimethylphenylsilyl endo enedione adduct (not shown); the latter was reduced in situ (CeCl3·7H2O, NaBH4) to afford mesodiol 23 as a white solid after purification by recrystallization from dichloromethane–hexanes (22.4 g, 50% yield from the total diene mass of 28.5 g, 67% yield from the amount of 22a present, mp 98–100 °C).
Two other conditions for retro-Diels–Alder fragmentation of the Michael–Claisen cyclization product 26 were also identified. Brief heating of 26 in diphenyl ether at 170 °C for 6 min provided the retro-Diels–Alder product 13 in 93% yield (Scheme 11). Also, heating 26, in the form of its hydrochloride salt, in hexafluoroisopropanol (HFIPA) at reflux for 12 h afforded the retro-Diels–Alder product 13 in 69% yield. Recovery of the silyl-substituted diene was not possible in the latter transformation, perhaps because protodesilylation occurred, though this speculation is not yet supported by experimental evidence.
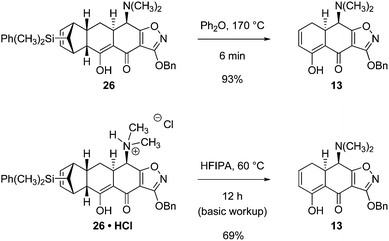 | ||
| Scheme 11 Alternative retro-Diels–Alder reaction conditions. | ||
With an improved sequence to the retro-Diels–Alder product 13 we had further opportunity to optimize the final three steps that transform this intermediate into AB enone 1 (Scheme 12). In the C12a-hydroxylation reaction, 3-(4-nitrophenyl)-2-(phenylsulfonyl)-oxaziridine was chosen as the stoichiometric oxidant in place of the original Davis oxaziridine in view of the reported greater thermal stability of the former33 and lithium tert-butoxide was used as base, in amounts as little as 20 mol%, in place of stoichiometric lithium hexamethyldisilazide as base. These adjustments to the protocol for C12a-hydroxylation led to an increased yield of product (14, 42.5 g, 85%, not purified). Our optimized protocol for epimerization employed a more concentrated aqueous solution of sodium dihydrogen phosphate and a higher reaction temperature; the ratio of the desired (4S)-epimer to the undesired (4R)-epimer increased to 12.8 to 1 with these modifications. Silylation of 15 proceeded as before (TBSOTf, 2,6-lutidine, CH2Cl2, 0 °C) with slight modifications in the workup which allowed for purification without resort to column chromatography. Thus, after silylation was complete, anhydrous methanol (9 mL) was added to quench any excess tert-butyldimethylsilyl trifluoromethanesulfonate; the volatile by-product, methyl tert-butyldimethylsilyl ether, was removed upon rotary evaporation. In addition, we found that 2,6-lutidine could be extracted selectively into pH 5 (0.1 M) aqueous sodium citrate buffer. After partial decolorization with charcoal, the AB enone 1 was obtained as an orange solid. Recrystallization from ethyl acetate provided a 1st crop of pure AB enone 1 as a white solid (31.6 g, mp 148–150 °C). Two additional crops of pure AB enone 1 were obtained upon further recrystallization from ethyl acetate–heptanes (2nd crop: 6.00 g, mp 148–150 °C; 3rd crop: 4.00 g, mp 149–151 °C). The optical rotation of the AB enone 1 ([α]24D + 148, c 1.13, CHCl3) confirmed that the product was of high optical purity.34 In the largest single-batch implementation of the optimized 5-step sequence outlined (beginning with the coupling of the cyclohexenone 25 and the isoxazole ester 3) we prepared 41.6 g of pure AB enone 1 in 35% yield overall. There were no chromatographic purification steps.
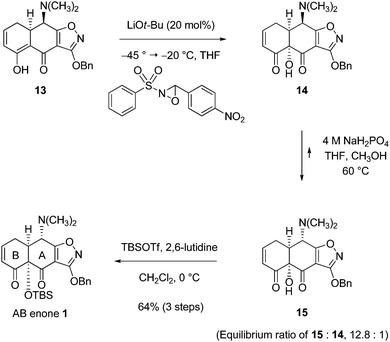 | ||
| Scheme 12 Synthesis of AB enone 1 (later reactions, optimized). | ||
Conclusions
The third- and fourth-generation routes to the AB enone 1 presented herein (employing cyclohexenones 2 or 25, respectively, and the isoxazole ester 3 as starting materials), like the second-generation route that preceded them, comprise 5-step synthetic sequences from a point of convergence that features the coupling of two simple starting materials, both readily prepared in large amounts. In the current work the two coupling components are of nearly equal complexity, as measured by the number of steps required to prepare each starting material. Asymmetry is introduced with the B-ring precursor and transferred via a highly diastereoselective Michael–Claisen cyclization to positions C4 and C4a and from these to position C12a by late-stage hydroxylation. In contrast, asymmetry is introduced in the second-generation route within the isoxazole precursor, at position C4, and this was relayed to positions C4a and C12a in a diastereoselective Diels–Alder cycloaddition reaction.3 The 5-step sequences of the second- and fourth-generation routes readily provide 40–50-g batches of crystalline AB enone 1 in comparable overall yields, using non-specialized laboratory apparatus. The fourth-generation route offers the advantage of many crystalline or solid intermediates, which permits synthesis of 1 without resort to chromatography (although a silica pad is employed in a recycling step). For kilogram-scale synthesis of fully synthetic tetracyclines, the fourth-generation route we present would require additional experimentation using appropriate and safe reaction vessels, however we believe that the current study provides a strong foundation for such research. Of greater importance in our ongoing efforts to discover new classes of tetracycline antibiotics is the fact that the fourth-generation route is more amenable to component modification for the preparation of structural variants of the AB enone 1, allowing modification of the tetracycline structure at positions such as C4, C4a, C5, and C12a, which heretofore have been relatively little explored because such compounds were largely inaccessible.Acknowledgements
Financial support from the National Institutes of Health (grant no. AI048825) is gratefully acknowledged. We also acknowledge Tetraphase Pharmaceuticals for predoctoral financial support (D.A.K.) and the FQRNT (Fonds quebecois sur la nature et les technologies) for postdoctoral fellowship support (A.D.). We thank Dr Shao-Liang Zheng for his guidance in X-ray data collection and structure determination, and Prof. David Evans and his research group for the use of their peristaltic pump.Notes and references
- M. G. Charest, C. D. Lerner, J. D. Brubaker, D. R. Siegel and A. G. Myers, Science, 2005, 308, 395 CrossRef CAS.
- C. Sun, Q. Wang, J. D. Brubaker, P. M. Wright, C. D. Lerner, K. Noson, M. Charest, D. R. Siegel, Y.-M. Wang and A. G. Myers, J. Am. Chem. Soc., 2008, 130, 17913 CrossRef CAS.
- J. D. Brubaker and A. G. Myers, Org. Lett., 2007, 9, 3523 CrossRef CAS.
- M.-C. Lasne, J.-L. Ripoll and J.-M. Denis, Tetrahedron Lett., 1980, 21, 463 CrossRef CAS.
- S. Takano, Y. Higashi, T. Kamikubo, M. Moriya and K. Ogasawara, Synthesis, 1993, 948 CrossRef CAS.
- K. Ogasawara, Pure Appl. Chem., 1994, 66, 2119 CrossRef CAS.
- H. Konno and K. Ogasawara, Synthesis, 1999, 1135 CrossRef CAS.
- For select examples, see: (a) S. Takano, M. Moriya, Y. Higashi and K. Ogasawara, J. Chem. Soc., Chem. Commun., 1993, 177 RSC; (b) S. Takano, M. Moriya and K. Ogasawara, Synlett, 1993, 601 CrossRef CAS; (c) S. Takano, Y. Higashi, T. Kamikubo, M. Moriya and K. Ogasawara, J. Chem. Soc., Chem. Commun., 1993, 788 RSC; (d) S. Takano, T. Kamikubo, M. Moriya and K. Ogasawara, Synthesis, 1994, 601 CrossRef CAS; (e) M. Shimizu, T. Kamikubo and K. Ogasawara, Tetrahedron: Asymmetry, 1997, 8, 2519 CrossRef CAS; (f) T. Kamikubo, M. Shimizu and K. Ogasawara, Enantiomer, 1997, 2, 297 CAS; (g) H. Nakashima, K. Hiroya, T. Taniguchi and K. Ogasawara, Synlett, 1999, 1405 CrossRef CAS; (h) G. Mehta and S. S. Ramesh, Tetrahedron Lett., 2004, 45, 1985 CrossRef CAS; (i) G. Mehta and H. M. Shinde, Chem. Commun., 2005, 3703 RSC; (j) T. J. Donohoe, P. D. Johnson, R. J. Pye and M. Keenan, Org. Lett., 2005, 7, 1275 CrossRef CAS.
- G. Stork and A. A. Hagedorn, J. Am. Chem. Soc., 1978, 100, 3609 CrossRef CAS.
- G. Stork, J. J. LaClair, P. Spargo, R. P. Nargund and N. Totah, J. Am. Chem. Soc., 1996, 118, 5304 CrossRef CAS.
- M. Oda, T. Kawaase, T. Okada and T. Enomoto, Organic Syntheses, 1996, 73, 253 CAS.
- A. P. Marchand, W. D. LaRoe, S. G. V. Madhava., S. C. Suri and D. S. Reddy, J. Org. Chem., 1986, 51, 1622 CrossRef CAS.
- K. E. Wilson, R. T. Seidner and S. Masamune, J. Chem. Soc. D, 1970, 213 RSC.
- S. Takano, M. Moriya, T. Kamikubo, K. Hiroya and K. Ogasawara, Tetrahedron Lett., 1993, 34, 8485 CrossRef CAS.
- M. Frey and V. Jager, Synthesis, 1985, 1100 CrossRef CAS.
- R. Riess, M. Schon, S. Laschat and V. Jager, Eur. J. Org. Chem., 1998, 473 CrossRef CAS.
- 1H NMR analysis of the unpurified reaction product also showed no evidence of any minor diastereomeric by-products.
- (a) A. Ichihara, Synthesis, 1987, 207 CrossRef CAS; (b) A. J. H. Klunder, J. Zhu and B. Zwanenburg, Chem. Rev., 1999, 99, 1163 CrossRef CAS; (c) B. Rickborn. In The Retro-Diels–Alder Reaction. Part I. C-C Dienophiles; Paquette, L. A. Ed.; Organic ReactionsVol. 52; John Wiley: New York, 1998; pp. 1–393 Search PubMed.
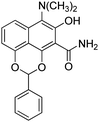
- An interesting by-product formed in varying yields during the high-temperature retro-Diels–Alder reaction is depicted below (the structure was determined by X-ray crystallographic analysis, see the Supporting Information for details†)
. - (a) L. H. Conover, J. J. Korst, K. Butler, R. B. Woodward and J. D. Johnston, J. Am. Chem. Soc., 1962, 84, 3222 CrossRef CAS; (b) J. J. Korst, J. D. Johnston, K. Butler, E. J. Bianco, L. H. Conover and R. B. Woodward, J. Am. Chem. Soc., 1968, 90, 439 CrossRef CAS.
- (a) H. Muxfeldt, G. Hardtmann, F. Kathawala, E. Vedejs and J. B. Mooberry, J. Am. Chem. Soc., 1968, 90, 6534 CrossRef CAS; (b) H. Muxfeldt, G. Haas, G. Hardtmann, F. Kathawala, J. B. Mooberry and E. Vedejs, J. Am. Chem. Soc., 1979, 101, 689 CrossRef CAS.
- (a) F. A. Davis, L. C. Vishwakarma, J. G. Billmers and J. Finn, J. Org. Chem., 1984, 49, 3241 CrossRef CAS; (b) F. A. Davis and B. C. Chen, Chem. Rev., 1992, 92, 919 CrossRef CAS.
- (a) A. P. Doerschuk, B. A. Bitler and J. R. D. McCormick, J. Am. Chem. Soc., 1955, 77, 4687 CrossRef CAS; (b) J. R. D. McCormick, S. M. Fox, L. L. Smith, B. A. Bitler, J. Reichenthal, V. E. Origoni, W. H. Muller, R. Winterbottom and A. P. Doerschuk, J. Am. Chem. Soc., 1957, 79, 2849 CrossRef CAS.
- M. M. U. S. Noseworthy. Patent 3,009,956, November 21, 1961.
- K. Alder, F. H. Flock and H. Beumling, Chem. Ber., 1960, 93, 1896 CrossRef CAS.
- (a) A. Ichihara, M. Kobayashi, K. Oda and S. Sakamura, Tetrahedron Lett., 1974, 15, 4231 CrossRef; (b) A. Ichihara, R. Kimura, K. Oda and S. Sakamura, Tetrahedron Lett., 1976, 17, 4741 CrossRef; (c) A. Ichihara, M. Kobayashi, K. Oda, S. Sakamura and R. Sakai, Tetrahedron, 1979, 35, 2861 CrossRef CAS; (d) A. Ichihara, K. Oda, M. Kobayashi and S. Sakamura, Tetrahedron, 1980, 36, 183 CrossRef CAS.
- P. Magnus, P. M. Cairns and J. Moursounidis, J. Am. Chem. Soc., 1987, 109, 2469 CrossRef CAS.
- P. A. Grieco and N. Abood, J. Chem. Soc., Chem. Commun., 1990, 410 RSC.
- S. Kotha, S. Banerjee, M. P. Patil and R. B. Sunoj, Org. Biomol. Chem., 2006, 4, 1854 CAS.
- (a) C. S. Kraihanzel and M. L. Losee, J. Am. Chem. Soc., 1968, 90, 4701 CrossRef CAS; (b) A. J. Ashe, J. Am. Chem. Soc., 1970, 92, 1233 CrossRef CAS; (c) S. McLean and G. W. B. Reed, Can. J. Chem., 1970, 48, 3110 CrossRef CAS.
- Y. Landais and L. Parra-Rapado, Eur. J. Org. Chem., 2000, 401 CrossRef CAS.
- A pure sample of cyclohexenone 25 was observed to undergo complete transformation to phenol when allowed to stand for 90 min at 23 °C on dry silica gel (2D-thin-layer chromatographic analysis).
- F. A. Davis, N. F. Abdul-Malik, S. B. Awad and M. E. Harakal, Tetrahedron Lett., 1981, 22, 917 CrossRef CAS.
- The optical rotation for AB enone 1 of 93% ee reported by Brubaker and Myers (Org. Lett., 2007, 9, 3523) was [α]23D −138 (c 0.52, CHCl3), which is in error. The correct rotation for AB enone 1 of 93% ee is [α]23D + 138 (c 0.52, CHCl3).
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data, copies of 1H and 13C NMR spectra, and X-ray crystallographic data for select compounds synthesized in this study can be found in the Supporting Information. CCDC reference numbers 826311–826313. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00303h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2011 |