Iron oligopyridine complexes as efficient catalysts for practical oxidation of arenes, alkanes, tertiary amines and N-acyl cyclic amines with Oxone†
Peng
Liu
,
Yungen
Liu
,
Ella Lai-Ming
Wong
,
Song
Xiang
and
Chi-Ming
Che
*
Department of Chemistry, State Key Laboratory of Synthetic Chemistry, and Open Laboratory of Chemical Biology of the Institute of Molecular Technology for Drug Discovery and Synthesis, The University of Hong Kong, Pokfulam Road, Hong Kong. E-mail: cmche@hku.hk
First published on 16th August 2011
Abstract
The iron complexes [Fe(qpy)](ClO4)2 (1) (qpy = 2, 2′![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6′, 2′′:6′′, 2′′′:6′′′, 2′′′′-quinquepyridine), [Fe2(spy)2](ClO4)4 (2) (spy = 2, 2′:6′, 2′′:6′′, 2′′′:6′′′, 2′′′′: 6′′′′, 2′′′′′-sexipyridine), [Fe2(septipy)2](ClO4)4 (3) (septipy = 2, 2′:6′, 2′′:6′′, 2′′′:6′′′, 2′′′′:6′′′′, 2′′′′′:6′′′′′, 2′′′′′′-septipyridine) and SBA-15-supported iron terpyridine complex (4) are efficient catalysts for the practical oxidation of alkenes, arenes, alkanes, tertiary amines and N-acyl cyclic amines using Oxone as the oxidant. With 1, 2, 3 or 4 as the catalyst, a variety of electron-rich and electron-deficient alkenes were oxidized to the corresponding epoxides in high isolated yields (up to 99%). The reaction of arenes with Oxone afforded quinones in up to 91% isolated yields. Selective C–H bond oxidation of saturated alkanes to alcohols/ketones and demethylation of tertiary amines to secondary amines were achieved in good product yields. For the oxidation of N-acyl cyclic amines, the ring-opened products were obtained in up to 75% isolated yield with up to 96% substrate conversion. All of these iron complexes can efficiently catalyse the oxidative C–C bond cross-coupling reaction of tertiary amines with NaCN in up to 86% isolated yield using (tBuO)2 as the terminal oxidant. The oligopyridine ligands were oxidatively robust and could be re-used by the addition of iron(II) salt to generate the iron catalystsin situ. The active SBA-15-supported terpy ligand could be re-used for the oxidative catalysis for five consecutive runs without significant loss of catalytic activity.
:6′, 2′′:6′′, 2′′′:6′′′, 2′′′′-quinquepyridine), [Fe2(spy)2](ClO4)4 (2) (spy = 2, 2′:6′, 2′′:6′′, 2′′′:6′′′, 2′′′′: 6′′′′, 2′′′′′-sexipyridine), [Fe2(septipy)2](ClO4)4 (3) (septipy = 2, 2′:6′, 2′′:6′′, 2′′′:6′′′, 2′′′′:6′′′′, 2′′′′′:6′′′′′, 2′′′′′′-septipyridine) and SBA-15-supported iron terpyridine complex (4) are efficient catalysts for the practical oxidation of alkenes, arenes, alkanes, tertiary amines and N-acyl cyclic amines using Oxone as the oxidant. With 1, 2, 3 or 4 as the catalyst, a variety of electron-rich and electron-deficient alkenes were oxidized to the corresponding epoxides in high isolated yields (up to 99%). The reaction of arenes with Oxone afforded quinones in up to 91% isolated yields. Selective C–H bond oxidation of saturated alkanes to alcohols/ketones and demethylation of tertiary amines to secondary amines were achieved in good product yields. For the oxidation of N-acyl cyclic amines, the ring-opened products were obtained in up to 75% isolated yield with up to 96% substrate conversion. All of these iron complexes can efficiently catalyse the oxidative C–C bond cross-coupling reaction of tertiary amines with NaCN in up to 86% isolated yield using (tBuO)2 as the terminal oxidant. The oligopyridine ligands were oxidatively robust and could be re-used by the addition of iron(II) salt to generate the iron catalystsin situ. The active SBA-15-supported terpy ligand could be re-used for the oxidative catalysis for five consecutive runs without significant loss of catalytic activity.
Introduction
The development of sustainable metal catalysts for selective organic oxidation, especially for C–H bond oxidation, is of paramount importance in organic synthesis.1 In the literature, most of the metal-catalysed organic oxidation reactions with practical interests often involve transition metal catalysts, such as those of Cr, Ru and Os. The conventional metal-based oxidants, such as CrO42−, [Pr4N][RuO4] and OsO4, display high activity but they are not bio-compatible. In the context of developing sustainable catalysis for organic synthesis, inexpensive and environmentally friendly catalytic oxidation reactions using cheap and nontoxic oxidants, such as H2O2,2a O22b or Oxone, are appealing, as the by-product is water or sulphate. Recently, iron-based oxidative catalysis has been proven to have practical applications. White reported the application of iron-catalysed aliphatic C–H bond oxidation in the modification of natural products.3 We have recently reported the iron-catalysed alkene epoxidation and aziridination of alkenes with practical interest.4b Using H2O2 as the terminal oxidant, Beller5 reported alkene epoxidation with a combination of iron(III) chloride and pyridine-2,6-dicarboxylic acid as the catalyst. Compared to other metals, iron is naturally abundant and non-toxic. The pivotal role played by iron-containing enzymes, such as cytochrome P450,6methane monooxygenase (MMO)7 and Rieske dioxygenases,8 in biological oxidative degradation is well-documented. A variety of synthetic non-heme iron complexes have recently been developed for bioinspired organic oxidations.9–12 For example, Que9 reported iron complexes containing tetradentate N4 ligands, such as [Fe(BPMEN)(NCCH3)2]2+ (BPMEN = N,N′-dimethyl-N,N′-bis(2-pyridylmethyl)-1,2-diaminoethane), [Fe(TPA)(NCCH3)2]2+ (TPA = tri(2-pyridylmethyl)amine) and [Fe(Me2PyTACN)]2+ (Me2PyTACN = 1-(2-pyridylmethyl)-4,7-dimethyl-1,4,7-triazacyclononane), all of which could catalyse alkene epoxidation, cis-dihydroxylation of alkenes and alkane hydroxylation. Ribas and Costas10 reported stereospecific C–H oxidation with [Fe(mcpp)]2+ (mcpp = N,N′-dimethyl-N,N′-bis[[5,6,7,8-tetrahydro-7,7-dimethyl-6,8-methanoisoquinolin-3-yl]methyl]-1,2-cyclohexanediamine) as the catalyst. Stack11 demonstrated that [((phen)2(H2O)Fe)2(μ-O)](ClO4)4 is an efficient catalyst for alkene epoxidation using peracetic acid as the oxidant. Que9 and Nam12 have elegantly characterized the structures and reactivities of non-heme iron(IV)-oxo complexes, both by X-ray crystallography and quantum mechanical calculations. Thus far, the practical application of non-heme iron-catalysed organic oxidation is hampered by low product selectivity (often attributed to the generation of free hydroxyl radicals) and a requirement of using a large excess of substrate(s) relative to the oxidant.In our endeavour to develop practical iron-catalysed organic reactions,4 we found that oligopyridines, an example of which is Cl3-tpy (Cl3-tpy = 4,4′,4′′-trichloro-2,2′:6′,2′′-terpyridine),4b are robust ligands that can be used to develop iron oxidative catalysis with practical interest. Herein, we describe that the iron oligopyridine complexes [Fe(qpy)](ClO4)2 (1) (qpy = 2, 2′:6′, 2′′:6′′, 2′′′:6′′′, 2′′′′-quinquepyridine), [Fe2(spy)2](ClO4)4 (2) (spy = 2, 2′:6′, 2′′:6′′, 2′′′:6′′′, 2′′′′:6′′′′, 2′′′′′-sexipyridine), [Fe2(septipy)2](ClO4)4 (3) (septipy = 2, 2′:6′, 2′′:6′′, 2′′′:6′′′, 2′′′′:6′′′′, 2′′′′′:6′′′′′, 2′′′′′′-septipyridine) and SBA-15-supported iron terpyridine complex (4) (Fig. 1) are efficient catalysts for the oxidation of alkenes, arenes, alkanes, tertiary amines and N-acyl cyclic amines, and for the oxidative cross-coupling reactions of tertiary amines with NaCN.
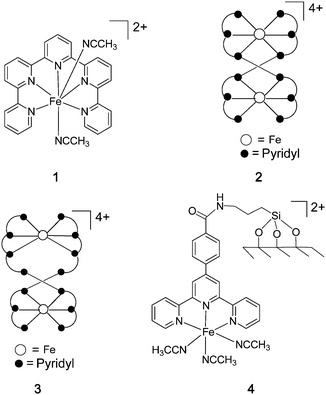 | ||
| Fig. 1 The structures of 1–4. | ||
Results and discussion
Synthesis and characterization of iron oligopyridine complexes
The iron oligopyridine complexes 14a and 44e were synthesized according to literature procedures. Following similar procedures, we prepared complexes 2 and 3 (see ESI† for details), both of which have been characterized by X-ray crystal structure determination (CCDC 818977 for 2, 818978 for 3). The crystal structure of 14a reveals a 7-coordinated iron complex. Both 2 and 3 are binuclear complexes with a helical structure (Fig. 2). Complex 2 crystallized as 2·CH3CN·1.5H2O. It contains two iron atoms coordinated with two spy ligands, similar to that of the cadmium–sexipyridine complex.13 The two ligand strands of spy are separated into two tridentate binding domains and coordinate to two Fe atoms lying on the helical axis. Similar to the structure of 2, the two ligand strands of septipy of 3 are also separated into two tridentate binding domains and coordinate to two Fe atoms. The C–C bonds connecting the two tridentate terpyridine subunits in complexes 2 and 3 are oriented by an angle of 68–70° and 23.07–37.01°, respectively. The average Fe–N(pyridine) distances of 1 and 2 are 2.28 Å and 2.113 Å, respectively. These values are typical for high spin Fe(II) complexes.14 Among the 14 nitrogen atoms of the two septipy ligand strands, except N4 and N11, 12 nitrogen atoms (each septipy ligand strand contributes 6 nitrogen atoms) are coordinated to Fe(II) in complex 3. Compared with the Fe–N distances of 2.09–2.34 Å in 3, the distances of Fe1–N4 (2.884 Å), Fe1–N11 (3.377 Å), Fe2–N4 (3.355 Å) and Fe2–N11 (2.838 Å) are much longer than that of a Fe–N single bond. The Fe–Fe distances in complexes 2 and 3 are 4.652 Å and 4.862–5.130 Å, respectively. These distances are considerably longer than twice the atomic radius of an iron atom and, hence, direct Fe–Fe interaction is precluded. The cyclic voltammograms of 1–3 in 0.1 M nBu4NPF6 CH3CN solution show a reversible oxidation couple at +0.5, 0.81 and 0.73 V (vs. Cp2Fe+/0), respectively, which is attributed to the oxidation of Fe(II) to Fe(III). The UV-visible absorption spectrum of 2 in CH3CN reveals two intense absorption bands with λmax at 320 and 552 nm, respectively. The UV-visible absorption spectrum of 3 in CH3CN features an intense absorption band at 306 nm and a weaker band at 532 nm. The intense absorption band is tentatively assigned to intra-ligand charge transfer transitions, while the less intense band is assigned to metal-to-ligand charge transfer transition(s) (MLCT). All of these three complexes are stable in the solid state and showed no sign of ligand dissociation in CH3CN/H2O (v/v = 1:9) for a period of 6 days under air, as revealed by UV-visible absorption spectroscopy. At room temperature, complexes 1–3 are paramagnetic, with μeff values of 4.94, 7.00, 7.20 B.M., respectively, corresponding to an S = 2 ground state.15 The 1H NMR spectra of 2 and 3 in CD3CN at room temperature display signals with chemical shifts in the −20 to +80 ppm and −10 to +100 ppm regions, respectively, revealing that the complexes are paramagnetic. ESI-MS analysis of a CH3CN solution of 2 revealed a prominent ion cluster peak at m/z 260.0374, with a peak separation of 0.25 mass unit, attributed to the [Fe2(spy)2]4+ formulation. For 3, a prominent ion cluster peak at m/z 298.5625 with a peak separation of 0.5 mass unit, which matches the simulated pattern with [Fe(septipy)]2+ formulation, was recorded under similar ESI-MS conditions.
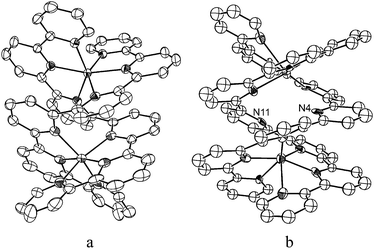 | ||
| Fig. 2 A side view of the complex cations of 2 (a) and 3 (b). | ||
Alkene epoxidation
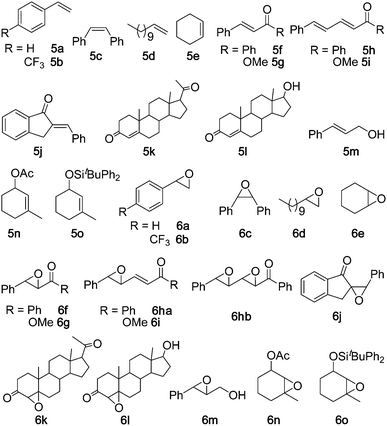
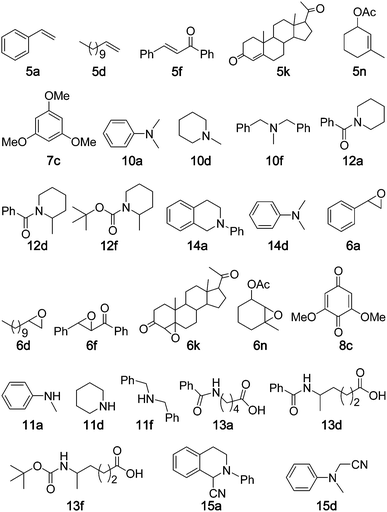
The epoxidation of alkenes using non-heme iron catalysts has been extensively studied; the overwhelming majority of previous studies were confined to electron-rich alkenes, such as cycloalkenes and styrenes. Reports on the efficient epoxidation of electron-deficient alkenes catalysed by non-heme iron complexes remain rare11 and involve the use of peracetic acid as oxidant. Following our studies on the epoxidation of a variety of alkenes with Oxone catalysed by [Fe(Cl3-tpy)2]2+,4b we set out to examine the epoxidation of styrene 5a catalysed by 1 under different reaction conditions (ESI†, Table S1). The best result of 92% substrate conversion with 82% yield of styrene oxide 6a was obtained in the presence of 5 mol% of 1 and NH4HCO3 (4.0 equiv.) with Oxone as the oxidant (1.3 equiv.) at room temperature for 2 h in aqueous CH3CN. Benzaldehyde (2%) was obtained as a minor side product. The epoxidation of styrene was pH dependent and the highest epoxide yield was obtained at pH 7. Using the protocol of “5 mol% of iron complex + Oxone + NH4HCO3”, complexes 1–3 were found to display comparable catalytic activities in the epoxidation of a wide range of alkenes (5a–o) including electron-deficient alkenes 5f–l (Table 1). Compared with the epoxidation of 5a–j, 5l, 5n and 5o catalysed by [Fe(Cl3-tpy)2]2+,4b the 1-catalysed counterparts (Table 1, entries 1–16, 18, 20, 23) gave comparable epoxide yields for 5a–h, 5n and 5o and higher epoxide yields for 5i, 5j and 5l (74–95% vs. 69–83%4b). In the epoxidation of α,β:γ,δ conjugated alkene 5i, both catalysts [Fe(Cl3-tpy)2]2+ and 1 led to γ,δ-monoepoxide 6i exclusively, in contrast to the selective formation of α,β-monoepoxide using [RuIV(2,6-Cl2TPP)Cl2] (H2(2,6-Cl2TPP) = meso-tetrakis(2,6-dichlorophenyl)porphyrin) as the catalyst and 2,6-dichloropyridine N-oxide (2,6-Cl2pyNO) as the terminal oxidant.16b The exclusive formation of a γ,δ-monoepoxide had been found in the epoxidation of ethyl sorbate (an analogue of 5i) with peracetic acid catalysed by a dinuclear non-heme iron complex, which also catalysed the epoxidation of 5g to give 6g in 86% yield.11 For cinnamyl alcohol 5m, its reaction with H2O2 catalysed by “iron(III) chloride + pyridine-2,6-dicarboxylic acid” gave 6m in up to 74% yield;5b,5c from the epoxidation of 5m with Oxone catalysed by 1, 6m was obtained in 93% yield (entry 19, Table 1). Substrate 5k (progesterone) has not previously been documented to be efficiently converted to epoxide 6k using a non-heme iron catalyst. A 65% yield of 6k (β isomer only), together with a 10% yield of another oxidation product of 5k, was reported for the oxidation of 5k with 2,6-Cl2pyNO catalysed by a ruthenium porphyrin in the presence of HBr.16a The epoxidation of 5k with Oxone catalysed by 1 afforded 6k as a mixture of α and β isomers with moderate α selectivity (α:β = 3:1) in 86% combined yield (substrate conversion: 90%) (Table 1, entry 17).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Product | Cat. | Conv. (%)b | Yield (%)c |
| a Substrate (0.2 mmol), catalyst (5 mol%), Oxone (0.26 mmol), NH4HCO3 (0.8 mmol), CH3CN (3 mL), H2O (2 mL), room temperature, 2 h. b Determined by GC or 1H NMR analysis of crude product. c Isolated yield based on starting substrate; isolated yield based on conversion is shown in brackets. d cis:trans ratio. e α:β ratio. f trans:cis ratio. | |||||
| 1 | 5a | 6a | 1 | 92 | 82(89) |
| 2 | 5a | 6a | 2 | 98 | 94(96) |
| 3 | 5a | 6a | 3 | 100 | 90(90) |
| 4 | 5b | 6b | 1 | 72 | 63(87) |
| 5 | 5c | 6c | 1 | 85 | 78(92) (27:1)d |
| 6 | 5d | 6d | 1 | 78 | 72(92) |
| 7 | 5d | 6d | 2 | 85 | 80(94) |
| 8 | 5d | 6d | 3 | 61 | 52(86) |
| 9 | 5e | 6e | 1 | 100 | 90(90) |
| 10 | 5f | 6f | 1 | 99 | 94(95) |
| 11 | 5f | 6f | 2 | 100 | 99(99) |
| 12 | 5f | 6f | 3 | 99 | 92(93) |
| 13 | 5g | 6g | 1 | 100 | 91(91) |
| 14 | 5h | 6ha/6hb | 1 | 96 | 62/32(65/33) |
| 15 | 5i | 6i | 1 | 100 | 90(90) |
| 16 | 5j | 6j | 1 | 100 | 95(95) |
| 17 | 5k | 6k | 1 | 90 | 86(96) (3:1)e |
| 18 | 5l | 6l | 1 | 82 | 74(90) (3:1)e |
| 19 | 5m | 6m | 1 | 100 | 93(93) |
| 20 | 5n | 6n | 1 | 78 | 64(82) (7:1)f |
| 21 | 5n | 6n | 2 | 88 | 84(96) (7:1)f |
| 22 | 5n | 6n | 3 | 87 | 78(90) (7:1)f |
| 23 | 5o | 6o | 1 | 70 | 55(78) (12:1)f |
Arene oxidation
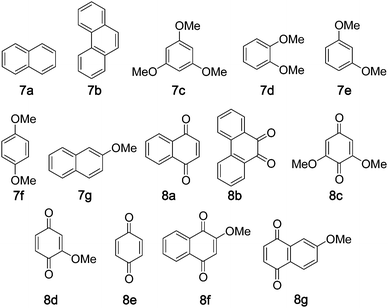
The oxidation of arenes to quinones is an important organic transformation due to the unique biological activities and synthetic applications of quinones.16,17 Biologically, arenes can be metabolized in vivo by cytochrome P-450 to form quinones.18 From a chemical perspective, the preparation of quinones by direct oxidation of arenes would be an advanced synthetic method. The most commonly employed procedures for the oxidation of arenes require an excess of metal-based oxidant, such as CrO3, which pose a severe problem to the environment.19 In the literature, iron-porphyrin complexes20b–d and non-heme iron complexes20a,20e have been reported to catalyse arene oxidation. Using the protocol “5 mol% of iron complex + Oxone + NH4HCO3”, naphthalene 7a was oxidized to 8a in 12% yield with 20% substrate conversion at 80 °C, while 8a was obtained in 28% yield and with 40% substrate conversion when using Oxone without NH4HCO3 (Table 2, entry 1). It was found that 1 underwent demetalation in the course of the reaction, but it could be easily regenerated by the addition of a new batch of Fe(ClO4)2·6H2O into the reaction mixture. So after every 6 h, new batches of Fe(ClO4)2·6H2O (5 mol%) and Oxone (1.2 equiv.) were added into the reaction mixture and 8a could be obtained in overall 47% yield and 72% substrate conversion for two repeated runs (Table 2, entry 1). The protocol was optimized as “5 mol% of iron complex + Oxone, add new batches of Fe(ClO4)2·6H2O (5 mol%) and Oxone (1.2 equiv.) into the reaction mixture every 6 h for two repeated runs”. Complexes 1–3 were found to display comparable catalytic activities in the arene oxidation. All the substrates 7a–g, except 7b, gave 1,4-quinones as the oxidation product. The previously reported oxidation of 7c with H2O2 catalysed by K3Fe(CN)6 gave 8c in up to 79% yield.20a When 7c was oxidized by Oxone using catalysts 1–3, 8c was obtained in 83–91% yields (substrate conversions: 85–92%) (Table 2, entries 5–7). Substrates 7a, 7b and 7d–f were not included in the previous oxidation of arenes catalysed by non-heme iron complexes.20a,20e The oxidation of 7d by “ruthenium porphyrin + 2,6-Cl2pyNO + HCl”17 and the oxidation of 7f by “iron porphyrin + Oxone”20b gave 8d and 8e in 11% and 32% yields, respectively. The 1-catalysed oxidation of 7a, 7b and 7d–f with Oxone afforded 8a, 8b, 8d and 8e in 45–83% yields (substrate conversions: 72–89%) (Table 2, entries 1, 4, 8–10).|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Product | Cat. | Conv. (%)b | Yield (%)c |
| a Substrate (0.2 mmol), catalyst (5 mol%), Oxone (0.24 mmol), CH3CN (3 mL), H2O (2 mL), 80 °C, 6 h; Fe(ClO4)2·6H2O (5 mol%) and Oxone (0.24 mmol) were added every 6 h for two repeated runs. b Determined by 1H NMR of crude product. c Isolated yield based on starting substrate; isolated yield based on conversion is shown in brackets. d After 6 h the reaction was quenched. | |||||
| 1 | 7a | 8a | 1 | 72 | 47(65) |
| 40d | 28(69)d | ||||
| 2 | 7a | 8a | 2 | 53 | 33(62) |
| 3 | 7a | 8a | 3 | 61 | 37(60) |
| 4 | 7b | 8b | 1 | 72 | 45(62) |
| 5 | 7c | 8c | 1 | 92 | 91(99) |
| 6 | 7c | 8c | 2 | 90 | 82(91) |
| 7 | 7c | 8c | 3 | 85 | 83(98) |
| 8 | 7d | 8d | 1 | 89 | 83(93) |
| 9 | 7e | 8d | 1 | 86 | 79(92) |
| 10 | 7f | 8e | 1 | 89 | 69(78) |
| 11 | 7g | 8f/8g | 1 | 68 | 29/26(42/38) |
Alkane oxidation
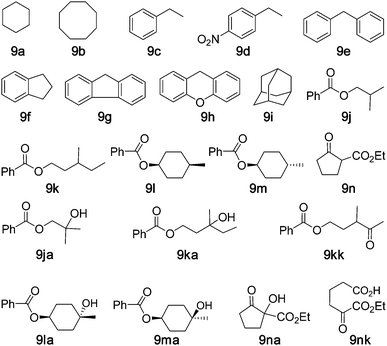
The selective oxidation of saturated C–H bonds of hydrocarbons is a major challenge in synthetic organic chemistry.21 Studies by Barton, also known as GIF Chemistry,22 and by Que23a,23b and Feringa23b,23d revealed that non-heme iron complexes can catalyse saturated C–H bond oxidation using oxygen, hydrogen peroxide or peracid as the oxidant. Cycloalkanes, such as cyclohexane and adamantane, are the most studied substrates in the alkane oxidation catalysed by non-heme iron complexes.23c,23e Recently, the works reported by White3 highlight the practicality of using unactivated saturated hydrocarbons as building blocks in the modification of natural products. Since the completion of this work, Nam12g reported a reactive non-heme iron(IV)-oxo complex that reacted with cyclohexane with a second order rate constant of 2.5 × 10−1 at −40 °C. Using our protocol of “5 mol% of iron complex + Oxone + NH4HCO3” for the oxidation of cyclohexane, the turnovers of cyclohexanol and cyclohexanone were 4.2 and 1.5, respectively. When the protocol was changed to “5 mol% of iron complex + Oxone”, the turnovers of cyclohexanol and cyclohexanone increased to 3.2 and 8.0 at 80 °C, respectively. Using the protocol for arene oxidation, i.e. “5 mol% of iron complex + Oxone, add new batches of Fe(ClO4)2·6H2O (5 mol%) and Oxone (1.2 equiv.) into the reaction mixture every 6 h for two repeated runs”, the turnovers of cyclohexanol and cyclohexanone were 3.6 and 12.0 for the oxidation conducted at 80 °C (Table 3, entry 1). Using complex 2 or 3 as the catalyst under similar reaction conditions, turnovers of 2.8, 3.1 for cyclohexanol and 9.2, 13.3 for cyclohexanone were observed, respectively (entries 2 and 3). Using complex 1 as the catalyst, the oxidation of hydrocarbons 9b–f afforded the corresponding ketones with yields up to 55% and with the ketone/alcohol (K/A) ratios up to 25 (entries 4–8). Fluorene (9g) and xanthene (9h) were exclusively oxidized to fluorenone and xanthenone in 81% and 85% yields, respectively (entries 9 and 10). Adamantane (9i) gave 1-adamantanol (26% yield) as the major product and 2-adamantanol (12% yield) and 2-adamantanone (4% yield) as minor products (entry 11), with a 3°/2° selectivity (per C–H bond) of 4.8, which is comparable to that reported for the oxidation with H2O2 catalysed by non-heme iron complexes.23c,23e White's work3b demonstrated the tolerance of various functional groups, including the ester group (and its electronic effect on site selectivity), in the C–H oxidation with H2O2 catalysed by a non-heme iron complex. We therefore examined substrates 9j–m, the hydroxylation of which catalysed by metal complexes, to the best of our knowledge, has not been reported before. The 1-catalysed hydroxylation of 9j–m with Oxone also predominantly occurred at 3° C–H bonds, with the ester group being well-tolerated. Oxidation products at 1° C–H bonds have not been detected. Isobutyl benzoate (9j) gave the tertiary alcohol (9ja) as the oxidation product in 53% yield with 73% conversion (entry 12), whereas 3-methylpentyl benzoate (9k) gave the tertiary alcohol (9ka, 33% yield) and ketone (9kk, 18% yield) with 71% conversion (entry 13). Cis- and trans-4-methylcyclohexyl benzoates (9l and 9m) were exclusively oxidized to give the tertiary alcohols4-hydroxyl-4-methylcyclohexyl benzoates (9la and 9ma) in 45% and 19% yields with retention of stereochemistry, respectively (entries 14 and 15). The oxidation of β-ketoester 9n was also examined, which gave hydroxylation product 9na in 67% yield (another product 9nk was obtained in 14% yield). A 90% yield of 9na was previously reported in the iron-catalysed oxidation of 9n with H2O2.23f|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | Cat. | Conv. (%)b | Yield (%)c | ||
| Alcohol (A) | Ketone (K) | K/A | ||||
| a Substrate (0.2 mmol), catalyst (5 mol%), Oxone (0.24 mmol), CH3CN (3 mL), H2O (2 mL), 80 °C, 2 h; Fe(ClO4)2·6H2O (5 mol%) and Oxone (0.24 mmol) were added every 6 h for two repeated runs. b Determined by GC or 1H NMR analysis of crude product. c Isolated yield based on starting substrate; isolated yield based on conversion is shown in brackets. d Turnover of the product. e Isolated yield of 1-adamantanol. f Isolated yield of 2-adamantanol. | ||||||
| 1 | 9a | 1 | — | 3.6d | 12.0d | 3.3 |
| 2 | 9a | 2 | — | 2.8d | 9.2d | 3.3 |
| 3 | 9a | 3 | — | 3.1d | 13.3d | 4.3 |
| 4 | 9b | 1 | 72 | 11(15) | 39(54) | 3.6 |
| 5 | 9c | 1 | 63 | 5(8) | 41(65) | 8.1 |
| 6 | 9d | 1 | 58 | 6(11) | 42(72) | 6.5 |
| 7 | 9e | 1 | 72 | 2(3) | 55(76) | 25 |
| 8 | 9f | 1 | 73 | 10(14) | 42(58) | 4.1 |
| 9 | 9g | 1 | 93 | 0(0) | 81(87) | — |
| 10 | 9h | 1 | 100 | 0(0) | 85(85) | — |
| 11 | 9i | 1 | 56 | 26e/12f(46e/21f) | 4(8) | 0.12 |
| 12 | 9j | 1 | 73 | 53(72) | 0(0) | — |
| 13 | 9k | 1 | 71 | 33(46) | 18(26) | — |
| 14 | 9l | 1 | 65 | 45(69) | — | — |
| 15 | 9m | 1 | 31 | 19(61) | — | — |
| 16 | 9n | 1 | 100 | 67(67) | 14(14) | — |
To explore the practicality of 1-catalysed C–H bond oxidation, two substrates were chosen and the corresponding oxidation reactions were conducted in a 10 mmol scale (scale up 50-fold). As depicted in Scheme 1, the reactions were performed using the protocol of “5 mol% of iron complex + Oxone, add new batches of Fe(ClO4)2·6H2O (5 mol%) and Oxone (1.2 equiv.) into the reaction mixture every 6 h for two repeated runs”. Xanthene was oxidized to xanthone in 83% yields with 100% conversion. Tetrahydronaphthalene was oxidized to 3,4-dihydronaphthalen-1(2H)-one (49% yield), 1,2,3,4-tetrahydronaphthalen-1-ol (12% yield) and 4-hydroxy-3,4-dihydronaphthalen-1(2H)-one (26% yield) with 96% conversion (Scheme 1).
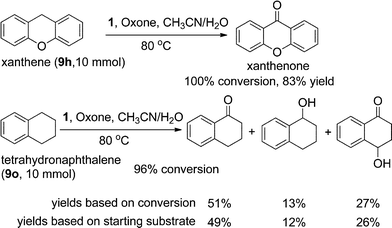 | ||
| Scheme 1 Scale-up (50-fold) of the oxidation reactions | ||
Amine oxidation: demethylation of tertiary amines
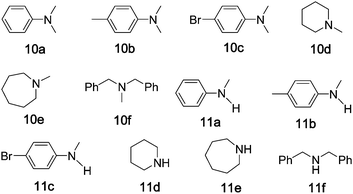
Due to the superior stability of N-methyl groups towards diverse reaction conditions, demethylation of tertiary N-methylamine remains a challenge in organic chemistry, especially for those complex molecules with functional groups.24 Some synthetic procedures have been developed to circumvent this problem. Murahashi reported the demethylation using ruthenium catalysts.25 Non-heme iron,26a,26biron porphyrin26c or iron powder26dcatalysts for the demethylation were also documented; demethylation of N,N-dimethylanilines 10a and 10b with oxygen using non-heme iron catalysts gave N-methylanilines 11a and 11b in up to 20% yields.26a,26b Using the protocol of “5 mol% of iron complex + Oxone”, the oxidation of N,N-dimethyaniline 10a catalysed by 1, 2 or 3 gave 11a in moderate product yields (51–62%) and with complete substrate conversion in each case (Table 4, entries 1–3). For the other dimethylanilines, 10b and 10c, 58% and 61% yields with complete substrate conversion were obtained, respectively (entries 4 and 5), though the yield of 10c is lower than that (84%) in the ruthenium-catalysed oxidation with tBuOOH.25a When cyclic tertiary amines (10d, 10e) were employed as substrates, the product yields were 72–82% (entries 6–9). Demethylation of dibenzylmethylamine 10f catalysed by 1, 2 or 3 gave 11f in 80–85% yields (entries 10–12). From the literature we have not found previous examples of metal-catalysed demethylation of 10d–f to 11d–f.|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Product | Cat. | Yield (%)b |
| a Substrate (0.2 mmol), Oxone (0.24 mmol), catalyst (5 mol%), CH3CN (3 mL), 5 h, room temperature. b Isolated yield based on substrate. | ||||
| 1 | 10a | 11a | 1 | 51 |
| 2 | 10a | 11a | 2 | 62 |
| 3 | 10a | 11a | 3 | 53 |
| 4 | 10b | 11b | 1 | 58 |
| 5 | 10c | 11c | 1 | 61 |
| 6 | 10d | 11d | 1 | 76 |
| 7 | 10d | 11d | 2 | 72 |
| 8 | 10d | 11d | 3 | 78 |
| 9 | 10e | 11e | 1 | 82 |
| 10 | 10f | 11f | 1 | 85 |
| 11 | 10f | 11f | 2 | 80 |
| 12 | 10f | 11f | 3 | 83 |
Amine oxidation: ring opening of N-acyl cyclic amines
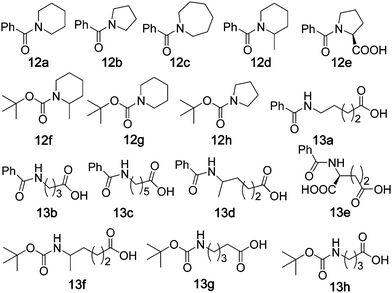
Direct oxidation of the C–H bonds of amides adjacent to a N atom is of importance. Only a few studies have been reported in this area. Nicolaou27 reported the oxidation of acyclic amides to imides using Dess–Martin periodinane as the oxidant. Higuchi28 reported that a “ruthenium porphyrin/pyridine N-oxide” protocol could catalyse the direct oxidation of N-acyl cyclic amines to give ring-opened terminal carboxylic acidsvia C–N bond cleavage. Using our protocol of “5 mol% of iron complex + Oxone, add new batches of Fe(ClO4)2·6H2O (5 mol%) and Oxone (1.2 equiv.) into the reaction mixture every 6 h for two repeated runs”, the oxidation of cyclic amide 12a gave 13a in 46% yield with 69% conversion at room temperature, and 59% yield with 90% conversion at 80 °C (Table 5, entry 1). Similarly, 13a was obtained in 58 and 57% yield when complexes 2 and 3 were employed as the catalyst (entries 2 and 3), respectively. Using complex 1 as the catalyst, all of these cyclic benzo- and tert-butyloxycarbamides (12a–h) were oxidized to terminal carboxylic acids 13a–h in 41–66% yields with high substrate conversions (86–96%). For α-substituted cyclic amides (12d–f), selective C–N bond cleavage occurred at the less-substituted carbons28 (entries 6–8). The 1-catalysed oxidation with Oxone, compared to the ruthenium-catalysed oxidation of 12a–f with 2,6-Cl2pyNO,28 gave a higher yield from 12c and comparable yields from 12a, 12b, 12d and 12f, albeit giving a lower yield from 12e.|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Product | Cat. | Conv. (%)b | Yield (%)c |
| a Substrate (0.2 mmol), catalyst (5 mol%), Oxone (0.24 mmol), CH3CN (3 mL), H2O (2 mL), 80 °C, 6 h; Fe(ClO4)2·6H2O (5 mol%) and Oxone (0.24 mmol) were added every 6 h for two repeated runs. b Determined by 1H NMR of crude product. c Isolated yield based on starting substrate; isolated yield based on conversion is shown in brackets. | |||||
| 1 | 12a | 13a | 1 | 90 | 59(65) |
| 2 | 12a | 13a | 2 | 93 | 58(62) |
| 3 | 12a | 13a | 3 | 95 | 57(60) |
| 4 | 12b | 13b | 1 | 90 | 63(70) |
| 5 | 12c | 13c | 1 | 92 | 55(60) |
| 6 | 12d | 13d | 1 | 96 | 75(78) |
| 7 | 12e | 13e | 1 | 86 | 41(48) |
| 8 | 12f | 13f | 1 | 93 | 66(71) |
| 9 | 12g | 13g | 1 | 90 | 50(56) |
| 10 | 12h | 13h | 1 | 92 | 50(54) |
Amine oxidation: oxidative C–C cross-coupling with NaCN
Transition metal-catalysed oxidative coupling of tertiary amines with nucleophiles is an efficient strategy for the synthesis of amino compounds, which are prevalent in bioactive natural products and in therapeutic drug molecules.29 Transition metal catalysts, such as CuBr,30iron complexes31 and RuCl3,32 are effective for this type of transformation reaction.33 In a previous work, we found that complex 2 is an efficient catalyst for the oxidative C–C cross-coupling of tertiary amines with indoles, pyrroles and alkynes.4e In this work, the oxidative C–C cross-coupling of tertiary amines with NaCN catalysed by 1, 2 or 3 and with (tBuO)2 as the terminal oxidant was demonstrated. As depicted in Table 6, tertiary amines, such as tetrahydroisoquinolines and N,N-dimethylanilines gave coupling products in good yields. In detail, the reactions of tetrahydroisoquinoline 14a, 14b with NaCN gave 15a, 15b with yields up to 86% (Table 6, entries 1–4). N-phenyl piperidine 14c gave 15c in 71% yield (entry 5). N,N-dimethylaniline 14d–f afforded 15d–f in up to 76% yield (entries 6–10). Similar or higher yields of 15a–f have been recently reported for the oxidation of 14a–f using the “FeCl2 + tBuOOH + Me3SiCN” protocol.31d The 1-catalysed reaction of N-methyl-N-ethyl aniline 14g with NaCN mainly occurred at the methyl group and gave 15g in 63% yield (entry 11), like the oxidation of 14g to 15g using the “iron phthalocyanine + H2O2 + NaCN”31e protocol.|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Product | Cat. | Yield(%)b |
| a Substrate (0.2 mmol), NaCN (0.24 mmol), catalyst (5 mol%), (tBuO)2 (0.4 mmol), CH3CN (3 mL), 80 °C, 12 h. b Isolated yield based on substrate. | ||||
| 1 | 14a | 15a | 1 | 82 |
| 2 | 14a | 15a | 2 | 86 |
| 3 | 14a | 15a | 3 | 80 |
| 4 | 14b | 15b | 1 | 79 |
| 5 | 14c | 15c | 1 | 71 |
| 6 | 14d | 15d | 1 | 76 |
| 7 | 14d | 15d | 2 | 72 |
| 8 | 14d | 15d | 3 | 68 |
| 9 | 14e | 15e | 1 | 63 |
| 10 | 14f | 15f | 1 | 61 |
| 11 | 14g | 15g | 1 | 63 |

Re-use of SBA-15-supported terpyridine ligand
In the course of the catalysis, demetalation was found for the iron oligopyridine complexes 1–3 and all of them could be easily regenerated by the addition of a new batch of Fe(ClO4)2·6H2O (5 mol%) to the reaction mixture. The ligands of the complexes 1–3 were found to be stable under oxidative conditions. For easy and convenient recovery of the ligand, we used the solid-supported SBA-15-terpy to react with Fe(ClO4)2·6H2O to give 4. The SBA-15-supported complex 4 was characterized by XRD, FT-IR, solid diffuse-reflectance and UV/Vis absorption spectroscopy.4e The loading of iron determined by atom absorption spectrophotometry was 1.9 wt %. Complex 4 could also be prepared in situ by the reaction of Fe(ClO4)2·6H2O with the ligand in a 1:1 ratio. Using epoxidation of chalone 5f as the model reaction, the recycling of 4 was studied. Upon completion of each reaction run, the initial purple color of complex 4 faded, revealing that demetalation took place. The SBA-15-supported terypridine ligand was recycled by filtration and treated with a new batch of Fe(ClO4)2·6H2O to regenerate the purple complex 4, which was used for subsequent reactions. High substrate conversions (up to 95%) and excellent product yields (86–96%) were obtained in five consecutive runs without significant loss of catalytic activity.
Some of the substrates described in previous sections were selected for the catalysis using SBA-15-supported complex 4 as the catalyst and Oxone as the terminal oxidant. As depicted in Table 7, all of the substrates gave the corresponding oxidation products with conversions and yields comparable to those obtained using complex 1, 2 or 3 as the catalyst.
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Product | Conv. (%)b | Yield (%)c |
| a Reaction conditions are similar to that in Table 2–6, but with complex 4 as the catalyst. b Determined by GC or 1H NMR analysis of the crude product. c Isolated yield based on starting substrate; isolated yield based on conversion is shown in brackets. d α:β ratio. e trans:cis ratio. | ||||
| 1 | 5a | 6a | 90 | 65(72) |
| 2 | 5d | 6d | 99 | 95(96) |
| 3 | 5f | 6f | 93 | 89(96) |
| 4 | 5k | 6k | 69 | 59(86) (3:1)d |
| 5 | 5n | 6n | 72 | 58(80) (7:1)e |
| 6 | 7c | 8c | 85 | 78(92) |
| 7 | 10a | 11a | 100 | 48(48) |
| 8 | 10d | 11d | 100 | 78(78) |
| 9 | 10f | 11f | 100 | 81(81) |
| 10 | 12a | 13a | 92 | 58(63) |
| 11 | 12d | 13d | 91 | 66(72) |
| 12 | 12f | 13f | 90 | 65(72) |
| 13 | 14a | 15a | 100 | 80(80) |
| 14 | 14d | 15d | 100 | 71(71) |
Mechanistic aspects
In the literature, iron-oxo9,12,34 complexes have been implicated in many non-heme iron catalysed oxidation reactions. Que and Nam first reported the X-ray crystal structure of non-heme iron(IV)-oxo complex [Fe(IV)(O)(TMC)(NCCH3)]2+ (TMC = 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane).34b To gain an insight into the mechanism of the oxidation reactions catalysed by 1, 2, 3 or 4, we studied the reaction intermediates using ESI-MS. Using CH3CN solutions of 1–3 in the presence of PhI(OAc)2 in a molar ratio of 1:4, prominent cluster peaks in positive-ion mode with m/z at 229.5355, 268.0576 and 306.5662, with a peak separation of 0.5 mass unit, were observed, which were assigned to [Fe(qpy)O]2+, [Fe(spy)O]2+ and [Fe(septipy)O]2+, respectively. Minor species, with m/z at 237.5347 and 314.5713, were also detected in the same reaction mixtures of 1 and 3, respectively. These two minor species are attributed to [Fe(qpy)O2]2+ and [Fe(septipy)O2]2+, respectively. All of the peak identifications were confirmed by their MS/MS CID spectra data. With consideration to our previous density functional theory calculations,4c,4f we propose that both monoxoiron(IV) (major) and dioxoiron(VI) (minor) species could be generated in the reaction mixtures under ESI-MS conditions.
Attempts to isolate [Fe(qpy)O]2+ and/or [Fe(qpy)O2]2+ were unsuccessful (see ESI† for {[Fe(qpy)(H2O)]2(μ-O)}(ClO4)4 obtained). To gain a further insight into the putative iron-oxo reactive intermediate(s), we performed competitive oxidation reactions using catalysts 1–3 for both the epoxidation of para-substituted styrenes and benzylic C–H bond oxidation of para-substituted ethyl benzenes. The log kY/kHvs. σ+ plots all gave a good linearity, with slope values (ρ+) of −0.60 to −0.67 for the epoxidation and −0.46 to −0.54 for the C–H bond oxidation (see ESI†). The kinetic isotopic effect (KIE) for the oxidation of cyclohexane by the protocol of “5 mol% of 1 + Oxone” was examined. A moderate primary KIE (kH/kD = 3.8 at 80 °C) was found. All of these findings are consistent with the formation of electrophilic iron-oxo species as reactive intermediate(s).9b,12b,12d,23
Conclusion
In summary, we have developed a simple protocol for practical oxidation of alkenes, arenes, alkanes, tertiary amines and N-acyl cyclic amines catalysed by iron oligopyridine complexes 1–3 with Oxone as the terminal oxidant and oxidative C–C bond coupling reactions with (tBuO)2 as the terminal oxidant. The operational principle is based on the generation of reactive iron-oxo intermediate(s). The SBA-15-supported ligand could be easily recycled to regenerate the active SBA-15-terpy iron complex for the oxidative catalysis for five consecutive runs without significant loss of catalytic activity. Although demetalation occurred during the oxidation reaction, the robust oligopyridine ligand could be re-used by adding new batch of iron salt.Acknowledgements
This work was supported by the Hong Kong Research Grants Council (HKU 1/CRF/08 and HKU 700810) and the Areas of Excellence Scheme established under the University Grants Committee of the Hong Kong Special Administrative Region, China (AoE 10/01P).Notes and references
-
(a) A. R. Dick and M. S. Sanford, Tetrahedron, 2006, 62, 2439 CrossRef CAS
; (b) K. Godula and D. Sames, Science, 2006, 312, 67 CrossRef CAS
-
(a) B. S. Lane and K. Burgess, Chem. Rev., 2003, 103, 2457 CrossRef CAS
-
(a) M. S. Chen and M. C. White, J. Am. Chem. Soc., 2004, 126, 1346 CrossRef CAS
-
(a) E. L.-M. Wong, G.-S. Fang, C.-M. Che and N. Zhu, Chem. Commun., 2005, 4578 RSC
-
(a) G. Anilkumar, B. Bitterlich, F. G. Gelalcha, M. K. Tse and M. Beller, Chem. Commun., 2007, 289 RSC
-
(a) B. Meunier, S. P. de Visser and S. Shaik, Chem. Rev., 2004, 104, 3947 CrossRef CAS
-
(a) M. Merkx, D. A. Kopp, M. H. Sazinsky, J. L. Blazyk, J. Müller and S. J. Lippard, Angew. Chem., Int. Ed., 2001, 40, 2782 CrossRef CAS
-
(a) D. T. Gibson and R. E. Parales, Curr. Opin. Biotechnol., 2000, 11, 236 CrossRef CAS
-
(a) K. Chen, M. Costas, J. Kim, A. K. Tipton and L. Que Jr, J. Am. Chem. Soc., 2002, 124, 3026 CrossRef CAS
-
(a) A. Company, L. Gómez, X. Fontrodona, X. Ribas and M. Costas, Chem.–Eur. J., 2008, 14, 5727 CrossRef CAS
- G. Dubois, A. Murphy and T. D. P. Stack, Org. Lett., 2003, 5, 2469 CrossRef CAS
-
(a) W. Nam, R. Ho and J. S. Valentine, J. Am. Chem. Soc., 1991, 113, 7052 CrossRef CAS
- E. C. Constable, M. D. Ward and D. A. Tocher, J. Am. Chem. Soc., 1990, 112, 1256 CrossRef CAS
- Y. Zang, J. Kim, Y. Dong, E. C. Wilkinson, E. H. Appelman and L. Que Jr, J. Am. Chem. Soc., 1997, 119, 4197 CrossRef CAS
- B. Weber and F. A. Walker, Inorg. Chem., 2007, 46, 6794 CrossRef CAS
-
(a) T. Iida, S. Ogawa, S. Miyata, T. Goto, N. Mano, J. Goto and T. Nambara, Lipids, 2004, 39, 873 CrossRef CAS
- T. Higuchi, C. Satake and M. Hirobe, J. Am. Chem. Soc., 1995, 117, 8879 CrossRef CAS
- O. Block, G. Klein, H.-J. Altenbach and D. J. Brauer, J. Org. Chem., 2000, 65, 716 CrossRef CAS
- S. Yamazaki, Tetrahedron Lett., 2001, 42, 3355 CrossRef CAS
-
(a) M. Matsumoto, H. Kobayashi and Y. Hotta, J. Org. Chem., 1985, 50, 1766 CrossRef CAS
-
B. Meunier, Ed., Biomimetic Oxidations Catalyzed by Transition Metal Complexes, Imperial College Press: Lodon, 2000 Search PubMed
-
(a) D. H. R. Barton and D. Doller, Acc. Chem. Res., 1992, 25, 504 CrossRef CAS
-
(a) C. Kim, K. Chen, J. H. Kim and L. Que, J. Am. Chem. Soc., 1997, 119, 5964 CrossRef CAS
-
(a) H. G. Stenmark, A. Brazzale and Z. Ma, J. Org. Chem., 2000, 65, 3875 CrossRef CAS
-
(a) S.-I. Murahashi, T. Naota and K. Yonemura, J. Am. Chem. Soc., 1988, 110, 8256 CrossRef CAS
-
(a) S. Murata, M. Miura and M. Nomura, J. Org. Chem., 1989, 54, 4700 CrossRef CAS
- K. C. Nicolaou and C. J. N. Mathison, Angew. Chem., Int. Ed., 2005, 44, 5992 CrossRef CAS
- R. Ito, N. Umezawa and T. Higuchi, J. Am. Chem. Soc., 2005, 127, 834 CrossRef CAS
- S.-I. Murahashi and D. Zhang, Chem. Soc. Rev., 2008, 37, 1490 RSC
-
(a) Z. Li, D. S. Bohle and C.-J. Li, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8928 CrossRef CAS
-
(a) S. Murata, K. Teramoto, M. Miura and M. Nomura, Bull. Chem. Soc. Jpn., 1993, 66, 1297 CrossRef CAS
-
(a) S.-I. Murahashi, T. Nakae, H. Terai and N. Komiya, J. Am. Chem. Soc., 2008, 130, 11005 CrossRef CAS
-
(a) A. J. Catino, J. M. Nichols, B. J. Nettles and M. P. Doyle, J. Am. Chem. Soc., 2006, 128, 5648 CrossRef CAS
-
(a) E. I. Solomon, T. C. Brunold, M. I. Davis, J. N. Kemsley, S. K. Lee, N. Lehnert, F. Neese, A. J. Skulan, Y.-S. Yang and J. Zhou, Chem. Rev., 2000, 100, 235 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and characterization of products. CCDC 818977 (complex 2), CCDC 818978 (complex 3) and CCDC 818979 ({[Fe(qpy)(H2O)]2(μ-O)}(ClO4)4) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre viahttp://www.ccdc.cam.ac.uk/data%5Frequest/cif. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00234a |
| This journal is © The Royal Society of Chemistry 2011 |