Operational calixarene-based fluorescent sensing systems for choline and acetylcholine and their application to enzymatic reactions†
Dong-Sheng
Guo
a,
Vanya D.
Uzunova
b,
Xin
Su
a,
Yu
Liu
*a and
Werner M.
Nau
*b
aDepartment of Chemistry, State Key Laboratory of Elemento-Organic Chemistry, Nankai University, Tianjin, 300071, P. R. China. E-mail: yuliu@nankai.edu.cn; Fax: (+86)2223503625
bSchool of Engineering and Science, Jacobs University Bremen, Campus Ring 1, D-28759, Bremen, Germany. E-mail: w.nau@jacobs-university.de; Fax: (+49)4212003229
First published on 23rd June 2011
Abstract
Electron-rich anionic calixarenes and resorcinarenes are known receptors for trimethylammonium-containing neurotransmitters, but the development of practical sensor applications has been impeded by the lack of suitable supramolecular sensing ensembles as well as the low selectivity and sensitivity of the macrocyclic cation-receptor hosts. The host–guest complexes between p-sulfonatocalix[n]arenes (n = 4–5) and the cationic aromatic fluorescent dye lucigenin (LCG) have been characterised by optical spectroscopic techniques, NMR, cyclic voltammetry, isothermal titration calorimetry, and X-ray crystallography. The dye is complexed with binding constants of the order of 107 M−1 and undergoes a strong static fluorescence quenching (factor 140) upon complexation as a consequence of exergonic electron transfer within the complex. LCG has been utilised in combination with p-sulfonatocalix[4]arene to set-up a refined reporter pair for label-free continuous real-time enzyme assays according to the supramolecular tandem assay principle. This affords product-selective tandem assays for amino acid decarboxylases with a one order of magnitude higher sensitivity and a 3 orders of magnitude lower host/dye concentration range, a convenient substrate-selective tandem assay for direct monitoring of choline oxidase, and a conceptually novel substrate-selective enzyme-coupled tandem assay for acetylcholinesterase. The applicability of the method to the measurement of enzyme-kinetic parameters, the screening for inhibitors of acetylcholinesterase, and the highly selective determination of absolute, low micromolar concentrations of both choline and acetylcholine by simple fluorescence measurements has been demonstrated. A domino tandem assay can be employed to measure the two analytes in the same sample. The described applications bypass problems related to the unselective binding of the macrocycle by coupling the signalling event with highly specific enzymatic transformations.
Introduction
Host–guest complexes between fluorescent dyes and calixarenes or resorcinarenes (Chart 1 and 2) have a rich history in the sensing of biological substances, particularly neurotransmitters of the acetylcholine and the carnitine type, as well as other organic analytes.1–17 The fluorescent dye is selected such that it shows a large change in fluorescence, ideally a strong quenching or enhancement, upon binding to the host. The general sensing principle is such that the addition of analyte causes dye displacement,18–20 which results in regeneration of the intrinsic fluorescence of the dye (Scheme 1). In theory, such sensing systems could afford a direct measure of absolute neurotransmitter concentrations as well as their fluctuations and thereby establish an extraordinarily important biological application. | ||
| Chart 1 Macrocycles employed for the sensing of acetylcholine and related quaternary ammonium ions, calixarenes 1–3 in this work. | ||
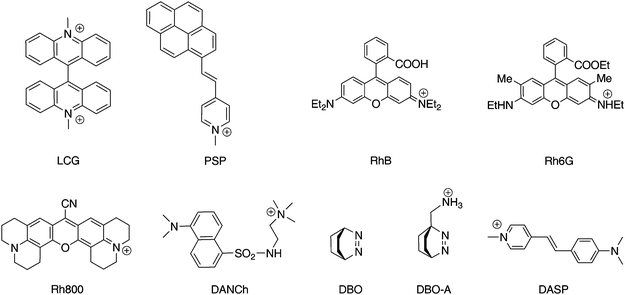 | ||
| Chart 2 Fluorescent dyes used in combination with the macrocycles in Chart 1 for the sensing of acetylcholine and related quaternary ammonium ions, LCG is used in this work. | ||
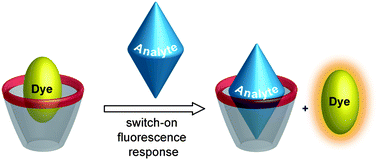 | ||
| Scheme 1 Fluorescence “switch-on” displacement assay for analyte sensing. | ||
A strong binding between the host and dye, a large fluorescence enhancement upon analyte binding, and operation in aqueous solution near neutral pH are the desired thermodynamic, photophysical, and solubility aspects which need to be successfully addressed in parallel to afford practically useful supramolecular sensing systems for neurotransmitters according to Scheme 1. Accordingly, much effort has been expended towards this end, and many combinations of different hosts (Chart 1) and dyes (Chart 2) have been investigated,1–17 which are detailed in the ESI†. Herein, we describe chemosensor applications based on novel host-dye reporter pairs composed of p-sulfonatocalix[4]arene (1), p-sulfonatocalix[5]arene (2), as well as p-sulfonatothiacalix[4]arene (3) as hosts (Chart 1) and lucigenin (LCG, N,N′-dimethyl-9,9′-biacridinium dinitrate), a common chemiluminescence agent,21–23 as the fluorescent guest (Chart 2).
All previously reported host-dye complexes4–6,9–17 show a sizable fluorescence response upon displacement of the fluorescent dye by the addition of neurotransmitters according to Scheme 1. While some of the most recently developed systems9,10,12,15,17 allow even operation in aqueous solution at physiological pH, the most important stumbling block towards practical application has remained the lack of specificity of the employed host molecules towards the biomolecular target analytes. Thus, while most of the macrocycles from Chart 1 show a certain selectivity, for example, they can differentiate well between acetylcholine (a cation) and glutamate (an anion), they interact very similarly with structurally related cationic analytes such as choline, acetylcholine, carbamoylcholine and L-carnitine.9–12 Even worse, numerous biologically abundant cations,7,24–26 biopolymers containing positively charged residues,8,13,14,26–30 and even simple salts as they are ubiquitous in biological systems31–35 bind significantly to the investigated calixarenes and resorcinarenes, which function as general cation receptors with varying but non-vanishing affinity for positively charged guests, and even neutral ones.36 In fact, Ballester and Rebek have gone so far as to state that such shallow macrocyclic receptors are useful for little more than recognizing a positive charge.37,38 This results in quantitatively unpredictable interferences due to interactions with variable metabolites in dependence on their concentration.39 For example, the presence of simple alkali and alkali-earth metal ions, as present in common electrolyte or buffer solutions (10–100 mM), markedly affects the binding of both dye and analyte with variations in binding constants by more than one order of magnitude not being uncommon.3,32–34 This unfortunate circumstance, namely, the lack of sufficiently high specificity, renders the determination of absolute concentrations of analytes in biological media according to the conventional displacement principle (Scheme 1) very difficult – if not practically impossible.
The determination of relative concentrations and concentration changes should nevertheless be feasible if the concentration of interfering metabolites and buffer ions remains constant. This exactly defines the idea of our present work in which we try to refine as well as redefine calixarene-dye sensor systems for biologically relevant applications, namely, for monitoring enzymatic reactions. Enzymatic catalysis constitutes a par excellence example for a reaction in which the relative concentration of a target analyte (product) changes dramatically (from zero to a finite value) while other concentrations remain constant. We have recently applied different combinations of macrocyclic hosts and fluorescent dyes (“reporter pairs”) in enzymatic reactions. In the resulting “supramolecular tandem assays”, the only prerequisite is a differential affinity of the macrocycle for substrate and product, which alone ensures a sizable fluorescence response once a suitable fluorescent dye has been identified.13,14,40–42 Our new reporter pairs composed of calixarenes 1–3 and LCG as a dye excel with respect to water solubility, affinity, and fluorescence response, and can be employed to set-up enzyme assays for acetylcholinesterase and choline oxidase. The assays also offer the possibility to screen for enzymatic inhibitors as well as to specifically determine, to return to the original incentive, absolute concentrations of both acetylcholine and choline.
Results and discussion
We have structured our experimental work such that we initially characterised the calixarene·LCG reporter pairs by using steady-state and time-resolved fluorescence, 1H NMR spectroscopy, X-ray crystallography, and cyclic voltammetry. An emphasis is placed on the mechanism of fluorescence quenching, which is critical for the design of practically useful reporter pairs. Subsequently, we have selected the concentration range with largest fluorescence response to monitor the activity of three different enzymes by employing the supramolecular tandem assay principle.13,14,40–42 We have first implemented the calixarene·LCG reporter pair in a previously developed tandem assay for lysine decarboxylase13,40,41 in order to expose the superior performance of our reporter pair (1·LCG) compared to the previously applied pair (1·DBO-A) with respect to substrate sensitivity as well as the working concentrations of host and dye.13 In a second step, we employed the 1·LCG reporter pair to set up a novel enzyme-coupled supramolecular tandem assay involving both acetylcholinesterase and choline oxidase.Binding constants, stoichiometry, and structure of host–guest complexes
Note beforehand that all employed calixarenes as well as the dye, LCG, are highly water-soluble, as required for the desired biological applications. Addition of calixarenes 1–3 (up to 9 μM) to very dilute aqueous solutions of LCG (1 μM) caused a strong fluorescence quenching (Fig. 1A), which was assigned, based on the associated Job's plot analyses (Fig. 1B and Fig. S4 in ESI†) as well as independent results obtained by UV-Vis as well as NMR spectroscopy (see below), isothermal titration calorimetry (Fig. S5 in ESI†), and X-ray crystallography (see below) to the formation of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 host–guest inclusion (precisely partial inclusion) complexes. The fluorescence depression (ca. factor of 140) is superior to most established fluorescent dye complexes in Chart 1 and 2, and ideal for the projected application, namely analyte sensing according to Scheme 1.
:1 host–guest inclusion (precisely partial inclusion) complexes. The fluorescence depression (ca. factor of 140) is superior to most established fluorescent dye complexes in Chart 1 and 2, and ideal for the projected application, namely analyte sensing according to Scheme 1.
![(A) Direct fluorescence titration of LCG (1 μM) with 1 (up to 8 μM) in 10 mM sodium perchlorate at pH 7, λex = 460 nm. The inset shows the associated titration curve, λem = 505 nm, and fit according to a 1 : 1 binding stoichiometry. (B) Job's plot for solutions of 1 and LCG in 100 mM phosphate buffer at pH 2, λex = 365 nm, λem = 505 nm, [1] + [LCG] = 8 μM.](/image/article/2011/SC/c1sc00231g/c1sc00231g-f1.gif) | ||
| Fig. 1 (A) Direct fluorescence titration of LCG (1 μM) with 1 (up to 8 μM) in 10 mM sodium perchlorate at pH 7, λex = 460 nm. The inset shows the associated titration curve, λem = 505 nm, and fit according to a 1:1 binding stoichiometry. (B) Job's plot for solutions of 1 and LCG in 100 mM phosphate buffer at pH 2, λex = 365 nm, λem = 505 nm, [1] + [LCG] = 8 μM. | ||
The binding geometry of LCG with calixarenes 1–3 was investigated by 1H NMR spectroscopy and X-ray crystallography. As can be seen from Fig. 2, all LCG protons exhibited fast exchange with upfield shifts in the presence of 1 equiv. calixarene, which are characteristic for the shielding effects of the macrocyclic aryl rings and support dye inclusion as the mode of interaction. With respect to the calixarene protons, the signals of the methylene bridges in 1 and 2 do not shift appreciably upon inclusion of LCG, but broaden to the baseline (or split into two sets of peaks). This is characteristic for a complexation-induced conformational rigidification of calixarenes.43 The conformational rigidification, evident from the NMR-spectral changes, is less pronounced for 2 than for 1, presumably due to its larger size and higher intrinsic flexibility.44–46 For calixarene 3, which has a more flexible cone compared to 147,48 and is more rigid compared to 2,45 NMR spectra cannot directly reveal any conformational change due to the lack of bridgehead –CH2– protons (Fig. S6 in ESI†).
 | ||
| Fig. 2 1H NMR spectra (all scales in δ (ppm)) of LCG (10 mM) with no additive or upon addition of 1 and 2 (10 mM) in deuterated sodium phosphate buffers at (A) pD 2 and (B) pD 7.2. The 1H NMR spectrum with 3 (10 mM) at pD 2 could not be recorded due to precipitation, while the spectrum at pD 7.2 can be found in Fig. S6 of the ESI†. ‘Ar–H’ and ‘–CH2–’ represent the aromatic and methylene bridge protons of the calixarenes, respectively. | ||
There are two acridine moieties in each LCG molecule, which assume a perpendicular conformation due to steric constraints. Hole-size arguments suggest that only one acridine moiety can be accommodated inside the cavity, a conjecture which could be experimentally verified for calixarene 2 by its crystal structure (Fig. 3). As can be seen, one of the acridine rings is included into the cavity of 2, while the other one “leans” onto the upper rim of the calixarene. This imposes a steric constraint towards the complexation of a second calixarene; indeed, experimentally, no indications for the formation of a 2:1 complex in solution were found. The detailed structural analysis reveals several discrete non-covalent interactions between 2 and LCG (Fig. S7 in ESI†), including two non-conventional hydrogen bonds (C52–H52⋯O14, 2.569 Å and 136.7°; C61–H61⋯O7, 2.689 Å and 129.6°), two C–H⋯π (C47–H47⋯ring of C9–C14, 3.118 Å and 177.2°; C49–H49⋯ring of C9–C14, 2.841 Å and 143.2°), and two π⋯π interactions (ring of C2–C7⋯ring of C43–C48, 3.945 Å; ring of C16–C21⋯ring of C43–C48, 4.283 Å), which rationalise further the high affinity between LCG and calixarenes 1–3.
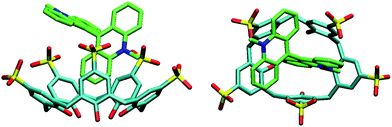 | ||
| Fig. 3 Crystal structure of the 2·LCG complex, side and top view. Note that the oxygen atoms of one of the sulfonato groups are delocalised over 6 positions; for clarity only 3 are shown. | ||
The association constants extracted from the fluorescence titrations, fitted according to a 1:1 stoichiometry, were very high, on the order of 107 M−1 at pH 7 (Table 1). They showed the known distinct trend towards lower values at acidic pH (cf. values in square brackets in Table 1),46,49,50 where one of the lower-rim phenolic hydroxyls becomes protonated,51 which diminishes both the negative charge status and cation receptor propensities.46,52 The drop in binding constants is most pronounced for 2 (and least pronounced for 3), which may reflect a higher rigidity of 2 at pH 2, due to a strengthening of the phenolic hydrogen-bonding network.53 Further inspection reveals that the binding constants are highest for 1 and lowest for 3, which is tentatively related to a preferred donor–acceptor π stacking for 1 due to its compactness, but a lower π stacking propensity for 3 due to its known lower electron-donor propensity. Apart from the rather small variations with structure and pH, the binding constants of LCG are 2 orders of magnitude larger than those of the neurotransmitters (e.g., 1.8 and 1.3 × 105 M−1 for acetylcholine and choline with 1),12 which presents an ideal condition54 for the desired highly sensitive (micromolar concentrations) monitoring of neurotransmitters within the biologically relevant range (see below).
| [Na+]/mMb | K a/(106 M−1)c | ||
|---|---|---|---|
| 1 | 2 | 3 | |
|
a Determined from fluorescence titrations by assuming a 1:1 stoichiometry; the corresponding binding constant fits are given in Fig. S2 in ESI†.
b Varying concentrations of sodium perchlorate, pH 7 (adjusted with NaOH).
c Error ±10%, unless stated explicitly.
d Values in square brackets are in 10 mM perchloric acid, pH 2 (see Fig. S1 in ESI†).
|
|||
| 0 | 28 ± 7 [2.5]d | 25 [0.7]d | 8.4 [0.45]d |
| 10 | 16 | 10 ± 2 | 0.44 ± 0.07 |
| 20 | 11 ± 2 | 5.3 | 1.9 |
| 50 | 0.40 | 1.5 | 0.56 |
| 100 | 0.07 | 0.50 | 0.17 ± 0.03 |
The binding constants were independently confirmed by ITC (see Fig. S5 and Table S1 in ESI†), which afforded comparable results (e.g., Ka = 2.5 × 107 M−1 for the 1·LCG complex at pH 7). As expected for the complexation of organic ammonium guests by calixarenes,28,45 the binding of 1–3 was found to be mainly enthalpically driven (ΔH ca. −42 kJ mol−1), accompanied by a negative entropic contribution (TΔS = −7 kJ mol−1) due to complexation-induced loss of conformational freedom for both macrocycle and host.
In order to assess potential interferences due to cations (cf. Introduction),3,32–34 we also performed titrations in solutions of varying ionic strength (0–100 mM NaClO4, Table 1).55 Indeed, the presence of sodium in concentrations typical for common buffers (50–100 mM) decreases the binding constant of LCG with calixarenes 1–3 significantly, by more than one order of magnitude. The data in Table 1 were further analysed (see Fig. S3 in ESI†) according to a competitive binding model (eq 8 in ESI†),32 which approximates the decrease of the observed (apparent) binding constant of a guest with increasing salt concentration. This analysis afforded binding constants for Na+ with 1–3 around 100 M−1, consistent with previous displacement titrations (85 M−1 for 1)32 and recent NMR measurements (100 M−1 for 1).35 The observed variations in binding constants with salts affect any sensing application involving calixarenes. For the enzyme-based methods described below, they affect, however, only the sensitivity of read-out (fluorescence differentiation), because the method employs the enzymes themselves for (salt-insensitive) neurotransmitter recognition.
Quenching mechanism
The exceptionally large fluorescence response upon complexation of LCG warrants detailed photophysical attention. As implied by the steady-state spectra (Fig. 1A), the fluorescence of LCG is quantitatively (>99%) statically quenched upon 1:1 complexation with calixarenes 1–3 due to the formation of a ground-state inclusion complex. Static quenching was confirmed by the upward curvature of the corresponding Stern–Volmer plots (inset of Fig. 4B). The formation of a ground-state complex was rigorously established by UV-Vis titrations, which revealed significant changes in the band shapes and intensities, as well as characteristic isosbestic points (Fig. 4A). The latter provide corroborative evidence for a two-state complexation model (uncomplexed form and 1:1 inclusion complex).
 | ||
| Fig. 4 (A) Absorption spectra of LCG (20 μM) titrated with up to 60 μM 2 in 100 mM sodium phosphate buffer, pH 7.2; (B) fluorescence spectra of LCG (1 μM) in the absence of additives (100%) and upon addition of 10 μM 1, 2, and 3 in 100 mM phosphate buffer, pH 2, λex = 368 nm. The inset shows the corresponding Stern–Volmer plot for calixarene 1; the deviation from linearity and upward curvature is characteristic for static quenching (fitted according to a 1:1 complexation model). | ||
Fig. 4B summarises the percentage of fluorescence quenching upon addition of identical amounts of calixarene (10 μM) to LCG (1 μM). The quenching efficiency is in accordance with the amount of complexed LCG and corresponds to the respective affinity at pH 2. As expected from the binding constants at pH 2 (values in square brackets in Table 1), 1 offers the highest quenching efficiency (89%), followed by the second strongest binding host 2 (75%), and the weakest binder, 3 (70%). Rigorous confirmation for static quenching was derived from time-resolved fluorescence measurements. The fluorescence lifetimes for free LCG show a weak pH dependence (19.4 and 16.5 ns for pH 2 and 7, respectively, Fig. S8 and Table S2 in ESI†), which is known.56 The addition of calixarenes greatly reduced the overall fluorescence intensity (indicative of a very short-lived species, the statically quenched inclusion complex), but left the fluorescence lifetime of the detectable component unaltered, within error (indicative of the absence of dynamic quenching of the residual uncomplexed LCG).
What is the mechanism of the static fluorescence quenching of LCG when complexed to calixarenes? With only two exceptions (the 6·DANCh and 5·DASP complexes), all fluorescent dyes from Chart 2 are quenched upon complexation by calixarenes (and resorcinarenes). Moreover, calixarenes are known to be good electron donors, e.g., they are involved as aryl units in cation–π and C–H π interactions.57–59 Conversely, LCG is a strong electron acceptor,21–23 such that a charge-transfer induced quenching in the complex appeared likely, a mechanism, which had been previously tentatively suggested for some other dyes from Chart 2.4,6,12,60 To investigate the tendency of charge transfer, we performed cyclic voltammetry measurements (Fig. 5 and Fig. S9 in ESI†) to determine the oxidation potentials (Eox) of the presumed electron donor species (calixarenes 1–3) as well as the reduction potential (Ered) of the electron acceptor LCG (Table 2). The data, and simple use of Rehm-Weller arguments,61 reveal that electron transfer is endergonic in the ground state of LCG by ca. +1.6 eV, but becomes strongly exergonic for singlet-excited LCG, where an additional −2.70 eV (corresponding to a fluorescence onset near 460 nm) are available from the electronic excitation energy (see Gibbs free energies of photoinduced electron-transfer in Table 2). To exclude the possibility that the electrochemical potentials undergo large shifts upon complexation, we also determined their values in the complexed state (Fig. 5 and Fig. S9 in ESI†, Table 2), but the complexation-induced electrochemical shifts were too small to offset the overall favourable excited-state electron-transfer thermodynamics. We therefore firmly conclude that LCG is quenched by the investigated calixarenes 1–3 by a full electron transfer. This efficient quenching mechanism rationalises the large fluorescence response upon complexation of LCG, which can be reversed by addition of competitive binders.
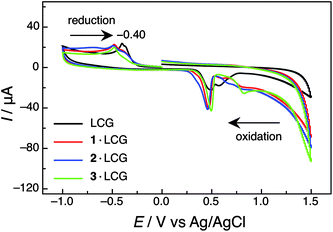 | ||
| Fig. 5 Cyclic voltammograms of LCG (500 μM) in the absence and presence of calixarenes 1–3 (500 μM) in 100 mM sodium phosphate buffer at pH 7.2. Note the electron reduction process of LCG, which is significantly shifted towards more negative values upon complexation by the calixarene macrocycles, due to the electrostatic stabilisation of the cationic guest. | ||
| Analyte | pH 2 | pH 7.2 | |||||
|---|---|---|---|---|---|---|---|
| E ox /Vb | E red /Vc | ΔGPET /eVd | E ox /Vb | E red /Vc | ΔGPET /eVd | ||
| a Determined by cyclic voltammetry with a scan rate of 100 mV s−1 at ambient temperature in 100 mM phosphate buffer (pH 2) or 100 mM sodium phosphate buffer (pH 7.2), cf. Fig. 5 and Fig. S9 in ESI†. b Estimated from the onset position of the irreversible oxidation waves; note that calixarenes undergo an irreversible oxidation, cf. ref. 62. c Peak potential. d Calculated from the simplified Rehm-Weller equation, ΔGPET = e(Eox − Ered) − E00, in which the Coulombic term has been omitted due to the large dielectric constant of the employed solvent, i.e., water. The singlet excitation energy of LCG (E00) was assumed to be pH-independent and estimated from the fluorescence onset at 460 nm as 2.7 eV. e Literature value in 50 mM Tris buffer is −0.38 V, cf. ref. 63. f Measured under conditions (500 μM calixarene and 500 μM LCG) of virtually quantitative (>96%) complexation of both species. | |||||||
| individual species | LCG | −0.41e | −0.40e | ||||
| 1 | 0.95 | −1.3 | 0.86 | −1.4 | |||
| 2 | 0.90 | −1.4 | 0.77 | −1.5 | |||
| 3 | 0.93 | −1.3 | 0.80 | −1.5 | |||
| complexesf | 1·LCG | 1.00 | −0.50 | −1.2 | 0.83 | −0.48 | −1.4 |
| 2·LCG | 0.91 | −0.49 | −1.3 | 0.80 | −0.50 | −1.4 | |
| 3·LCG | 0.93 | −0.45 | −1.3 | 0.82 | −0.48 | −1.4 | |
Comparison with established reporter pairs
The pertinent properties of the LCG reporter pairs designed for neurotransmitter sensing by fluorescent dye displacement applications are compared in Table 3 with those of existing host-dye systems. As can be seen, the novel calixarene·LCG sensing ensembles meet not only the desired water solubility and operation over a broad pH range, but show also an advantageous high affinity (larger than that of neurotransmitters, which serve as competitors).54 The large binding constant of LCG (see also ITC data in Table S1, ESI†) in comparison to the other dyes can be nicely accounted for by the advantageous goodness-of-fit (Fig. 3) and the numerous types of synergistic noncovalent interactions (see above). Additionally, LCG shows the largest fluorescence responses (more than 2 orders of magnitude variation) among the investigated dyes. This is a favourable combination of beneficial aspects not found for any of the alternative, previously documented, sensing systems (see Section 1.1. in ESI†).| Host·dye | I free/Ibounda | K a/(106 M−1) | Solvent (pH) | Ref. |
|---|---|---|---|---|
| a Fluorescence intensity of the uncomplexed dye divided by that of the complexed dye, extrapolated to quantitative complexation from direct host–guest titrations; this value corresponds to the maximum theoretical fluorescence change upon analyte addition. b Value extrapolated from direct host-dye titrations, e.g., Fig. 1A; pH- and salt-dependent (see Table 1). c Estimated from the maximal fluorescence regeneration upon analyte addition to the preformed host–guest complexes. | ||||
| 1·LCG | >140b | 28 | water or buffer (pH 2–8) | this work |
| 2·LCG | >140b | 25 | water or buffer (pH 2–8) | this work |
| 3·LCG | >140b | 8.4 | water or buffer (pH 2–8) | this work |
| 4·PSP | 2c | 0.23 | KOH/MeOH | 4 |
| 5·PSP | 5 | ∼0.006 | water/MeOH (pH 8) | 5 |
| 6·RhB | 5 | 0.14 | phosphate buffer (pH 6.6) | 6 |
| 6·Rh6G | 3.3 | 1.9 | phosphate buffer (pH 6.9) | 9 |
| 6·Rh800 | 3.3 | 0.45 | phosphate buffer (pH 7.2) | 10 |
| 6·DANCh | 0.6 | 0.33 | water (pH 6.9) | 11 |
| 7·PSP | 2 | 4.26 | phosphate buffer (pH 8) | 16 |
| 1·DBO | 10 | 0.001 | water or buffer (pH 2–8) | 12 |
| 1·DBO-A | 8c | 0.023 | water or buffer (pH 2–8) | 12–15 |
| 5·DASP | 0.02 | 0.1 | phosphate/MeOH (pH 7.2) | 17 |
Implementation in supramolecular tandem assays
We have recently introduced the supramolecular tandem assay principle (Scheme 2) as a convenient method to monitor enzymatic activity,13,14,40–42 to analyze absolute concentrations of substrates of enzymatic reactions,14 and also to quantify enantiomeric purity of substrates.40 The method makes use of an indicator displacement strategy with a fluorescent dye from a macrocycle for signalling (Scheme 1)18–20in combination with enzymes to achieve high selectivity. The role of the macrocycles is merely to differentiate between a substrate and a product (one of them has to bind at least a factor of 10 or more strongly than the other);14 a high selectivity in the supramolecular recognition process is not required. As the enzymatic reaction proceeds in the presence of the macrocycle-dye reporter pair, the chemical equilibrium is changed such that the fluorescent dye is either, as the reaction proceeds, displaced from the cavity (product-selective assay), or taken up into the macrocyclic cavity (substrate-selective assay). This results in a readily detectable fluorescence response, which can be employed to determine the enzymatic activity. Whether the fluorescence is switched-on or -off in the course of the assay depends on the photophysical properties of the dye and the macrocycles; for the investigated calixarenes, the dye is generally quenched in the complexed form (see quenching mechanism above and Table 3) such that the fluorescence responses specified in Scheme 2 result. | ||
| Scheme 2 Schematic representation of a product- and substrate-selective tandem assay involving fluorescent dyes and calixarenes as macrocycles. | ||
To adapt tandem assays to various enzymatic reactions, we have already explored reporter pairs composed of different fluorescent dyes and macrocycles, including cucurbiturils,13,14 cyclophanes and cyclodextrins,42 as well as one calixarene-based reporter pair (1·DBO-A).13 We are continuously working to expand the library of reporter pairs in order to be able to sense various types of enzymatic transformations. Calixarenes 1–3,64–66 similar to cucurbiturils67 and cyclodextrins,68,69 show only low cellular toxicity, which paves the way for potential biological applications. From those we have mainly focused on the 1·LCG reporter pair as a prototype of the investigated calixarene·LCG complexes, because both the LCG dye and the macrocycle 1 are commercially available and can be easily implemented for future sensing applications. The other two reporter pairs (2·LCG and 3·LCG) remain interesting alternatives should the reporter pair 1·LCG show short-comings for a particular application which we did not yet encounter.
In order to test the compatibility of the refined reporter pair 1·LCG, we have first compared its performance in a previously introduced product-selective supramolecular tandem assay for amino acid decarboxylases (EC 4.1.1.18)13,40,41 with that of the established reporter pair 1·DBO-A. The detailed assay results for lysine decarboxylase are found in the ESI† because they present only an optimisation, even if significant, of an already documented application.13 They demonstrate not only that the reporter pair 1·LCG can be principally applied in enzyme assays, but also that it outperforms the previously employed reporter pair 1·DBO-A in several respects. First, owing to the higher host-dye binding constant (Table 3), the employed dye and host concentrations could be decreased by 2 orders of magnitude, from 100 μM dye and 400 μM host for 1·DBO-A13 to 0.5 μM dye and 1.5 μM host for 1·LCG. Second, the substrate concentration could be lowered by one order of magnitude, from 500 μM for 1·DBO-A13 to 50 μM for 1·LCG. Both improvements are highly desirable from a sensitivity and economic as well as an interference point of view, particularly for potential applications in high-throughput screening for drug discovery.14
In the next stage, we built upon the known affinity of calixarene 1 for neurotransmitters and selected enzymes which accept those targets as substrates, namely acetylcholinesterase (EC 3.1.1.7) and choline oxidase (EC 1.1.3.17). They catalyze in an enzymatic reaction cascade the hydrolysis of the neurotransmitter acetylcholine to choline and the oxidation of choline to betaine, respectively (Scheme 3, top).
 | ||
| Scheme 3 Reactions catalyzed by acetylcholinesterase and choline oxidase and the corresponding (enzyme-coupled) tandem assays. | ||
Keeping an eye on potential biological applications, choline oxidase (ChO) is responsible for regulating the osmotic equilibrium in cells by maintaining the levels of water-soluble organic amines (choline and betaine).70,71 Genetic engineering has been aimed towards the regulation of ChO activity with the objective to develop plants with higher resistance towards osmotic stress (e.g., to improve frost resistance).72 The enzyme has also been tested in detergents for a continuous in situ release of H2O2 for bleaching combined with the fabric softener effect of the betaine quaternary ammonium ion.73 Surprisingly, while ChO has a broad range of applications, no direct method is available to assay its activity. Existing methods involve either derivatised substrates (Ellman method with thiocholine and 5-thio-2-nitro-benzoic acid as chromogenic additive),74 instrumentally-demanding oxygen consumption methods,75–77 or peroxidase-coupled methods. The latter probe the formation of H2O2 either by electrochemical methods,78,79 or by oxidative production of a chromophore.80–82
Acetylcholinesterase (AChE), on the other hand, is a key enzyme involved in the development of Alzheimer's dementia and efficient inhibitors are being sought as the best drugs for its treatment.83 The research on AChE inhibition started with the finding that neuronal degeneration in certain brain areas is connected with reduced levels of neurotransmitters.84,85 Therefore, AChE inhibitors are proposed as a way to suppress the acetylcholine neurotransmitter breakdown and delay the processes of mental degradation. This renders the screening of AChE inhibitors essential for the development of novel Alzheimer's therapeutics. Due to the high interest in the AChE activity and inhibition, several methods have been developed for monitoring its activity. They often involve sequences of reagents and enzymatic transformations,74,86 or complex chemiluminescent,87 radiolabelling88 or electrochemical measurements89 in order to obtain sizable responses. As can be seen, there is a high demand for convenient assays for the two neurotransmitter-related enzymes to ease the search for inhibitors, such that the use of label-free supramolecular tandem assays would open interesting possibilities. The simplicity of the method we describe in the following is compelling; it requires neither specialised instrumentation nor labelling agents, and is particularly attractive for inhibitor screening.
Competitive titrations with the substrates and products of the enzymatic reactions stand at the basis of the development of any tandem assays. The 1·LCG reporter pair was consequently first titrated with the various analytes (Fig. 6A), which revealed similar fluorescence enhancements upon addition of acetylcholine and choline. Thus, unfortunately, a direct tandem assay for the hydrolysis of acetylcholine to choline could not be set up, because both the substrate and product have almost the same binding affinity to calixarene 1 (ca. 1.0 × 105 M−1, Fig. 6A);9–12 this presents just another experimental manifestation of the low selectivity of 1.7–14,24–38 However, we realised that the enzymatic conversion of choline (substrate, strong competitor) to betaine (product, weak competitor) can be easily monitored with the 1·LCG reporter pair because this substrate/product pair shows a fluorescence differentiation up to a factor of ca. 3 (Fig. 6A). This corresponds to a difference in binding constants of a factor of 200 or more, which is evidently due to the change in charge status between these two metabolites. The observed substrate/product differentiation allowed us not only to set-up convenient substrate-selective tandem assays for ChO itself, but also for AChE, because the initial product choline could be converted in situ to the final product betaine (Scheme 3). The formation of betaine would then trigger the desired fluorescence response, which would give, provided that the relative enzyme concentrations were correctly selected from an enzyme-kinetic point of view, information on the kinetics of acetylcholine hydrolysis, changes in enzymatic activity (in the presence of inhibitors or activators), and, where desirable, the absolute concentration of acetylcholine. Note that the proposed tandem assay for acetylcholine needs also to be qualified as a substrate-selective one, because the (initial and intermediary) substrates acetylcholine and choline act as strong competitors, while betaine is the weak one. Since assays involving a sequence of enzymatic reactions are generally known as “enzyme-coupled” assays, we refer to the conceptually novel assay for AChE as a substrate-selective enzyme-coupled supramolecular tandem assay.
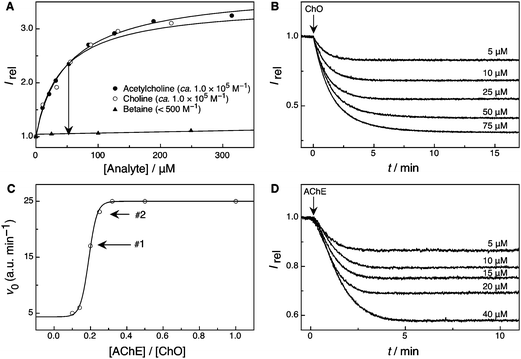 | ||
| Fig. 6 (A) Competitive fluorescence titrations of acetylcholine and its enzymatic products choline and betaine in the presence of LCG (100 nM) and 1 (180 nM) in 10 mM sodium phosphate buffer, pH 8 (λex = 410 nm, λem = 505 nm).90 The expected fluorescence response in the course of the enzymatic reaction is represented by an arrow. (B) Continuous fluorescent enzyme assay for ChO with the 1·LCG reporter pair (0.5 U ml−1 ChO, 5–75 μM choline, 0.7 μM LCG, and 2.0 μM 1 in 10 mM sodium phosphate buffer, pH 8, λex = 410 nm, λem = 505 nm). (C) Dependence of the initial reaction rates on the concentration of AChE (0.05–0.5 U ml−1) in the presence of a constant amount of 0.5 U ml−1 ChO, 40 μM acetylcholine, 700 nM LCG, and 2 μM 1 in 10 mM sodium phosphate buffer, pH 8 (λex = 410 nm, λem = 505 nm). Arrows #1 and #2 indicate the selected concentrations for the acetylcholine concentration dependence (Fig. 6D) and the inhibition experiments (Fig. 7). (D) Enzyme-coupled tandem assay for AChE with the 1·LCG reporter pair (0.1 U ml−1 AChE and 0.5 U ml−1 ChO, 5–40 μM acetylcholine, 180 nM LCG, and 450 nM 1 in 10 mM sodium phosphate buffer, pH 8, λex = 410 nm, λem = 505 nm). | ||
Because the assays according to Scheme 3 operate in the substrate-selective mode and afford a weakly binding product, a switch-off fluorescence response was expected when the assay was performed at a fixed concentration of substrate (e.g., 50 μM, arrow in Fig. 6A). In fact, the ChO assay worked exactly as projected (Fig. 6B, the concentration of ChO was selected to achieve conversion within 15 min) and showed the expected dependence of the enzyme kinetics on the selected substrate concentration. A KM value of 160 ± 10 μM (Fig. S12A in ESI†) was estimated from the resulting Lineweaver–Burk plot, on the same order of magnitude as the literature value of 530 ± 20 μM.77 The final plateau region also depended on (decreased with) the substrate concentration, because larger amounts of the more strongly binding substrate are capable of displacing a larger fraction of the dye at the outset of the enzymatic reaction. Gratifying to observe, the largest fluorescence decrease amounted to a factor of 2–3, as theoretically expected, cf. arrow in Fig. 6A.
Also the enzyme-coupled assay with AChE as a second enzyme and acetylcholine as a substrate worked very well, showing again the characteristic dependence of the plateau region with varying substrate concentration (Fig. 6D). The enzyme-kinetic parameters could also be extracted from these traces, but the relative concentrations of the two enzymes needed to be matched for the kinetic analysis to be quantitatively meaningful. In particular, we needed to ascertain that the first reaction step (hydrolysis of acetylcholine) was rate-determining. We consequently determined the initial reaction rates at the ChO concentration already optimised for the ChO assay, but varied the amounts of AChE (Fig. 6C). As can be seen, the initial reaction rate reached a plateau value at high acetylcholine concentrations, suggesting that at these concentrations the formation of betaine was rate-limited by ChO. At lower concentrations, the reaction rate decreased with concentration of AChE, and within a ratio range of 0.20 ± 0.05 an approximately linear decrease was observed. This is the kinetic regime in which the reaction rate is limited by AChE, and this is also the AChE concentration ratio (0.20, see arrow #1, Fig. 6C) selected for the investigation of the substrate dependence (Fig. 6D). Indeed, Lineweaver–Burk plots afforded KM = 70 ± 20 μM (Fig. S12B in ESI†) which is in very good agreement with the literature value of 90 μM).91 Note that the absolute fluorescence intensities for the AChE assay as well as the fluorescence response were slightly less (a factor of 2 decrease) than those obtained for the ChO assay. This was mainly due to the fact that we lowered the concentration of the reporter pair by one order of magnitude for the AChE assay. This allowed nanomolar concentrations of both dye and host to be used, demonstrating that the reporter pair can be truly used as a spectator in the enzymatic reactions. We have previously shown that this is a particularly important point for substrate-selective tandem assays in order not to significantly affect the effective concentration of substrate by macrocyclic complexation.14
It should be noted that the enzymatic activity of ChO and AChE was independently monitored by mass spectrometry (Fig. S13 in ESI†), which confirmed that the enzymatic reaction proceeded indeed on the same time scale as that observed in the fluorescence measurements. We also confirmed (by fluorescence) that the enzymes or any presence of hydrogen peroxide did not affect the fluorescence of the 1·LCG reporter pair (Fig. S14A,B in ESI†), and that the reporter pair did not affect the enzymatic reaction itself (control experiments by mass spectrometry, see Fig. S13 in ESI†). Finally, we also performed control experiments to show that no fluorescence response was obtained from acetylcholine in the absence of AChE (as it could occur if hydrolysis occurred otherwise) and that no significant response was obtained in the absence of ChO oxidase (i.e., when the reaction would halt at the choline stage, see Fig. S14C,D in ESI†).
Screening of inhibitors
To demonstrate the suitability of the enzyme-coupled tandem assay for the screening of inhibitors for acetylcholine, we focused on two known reversible competitive inhibitors: Tacrine and (–)-Huperzine A, where the former is an approved Alzheimer's drug and the latter is a potent dietary supplement for Alzheimer's patients.92 For the inhibition experiments, we selected the upper concentration ratio of AChE/ChO within the approximately linear range of the plot in Fig. 6C (0.25, see arrow #2), because the addition of competitive inhibitors was expected to apparently lower this value (by reducing the amount of active AChE). As can be seen, the addition of increasing amounts of the additives led to an efficient suppression of AChE activity, reflected in a steep decrease in initial reaction rates. Hill analysis of the inhibition plots14 afforded Ki values93 of 12 ± 1 nM for Tacrine and 76 ± 6 nM for (–)-Huperzine A (Fig. 7), which are in very good agreement with literature reports (10–25 nM for Tacrine94–96 and 14–175 nM for (–)-Huperzine A,95–97 obtained by colorimetric assays according to the Ellman method74).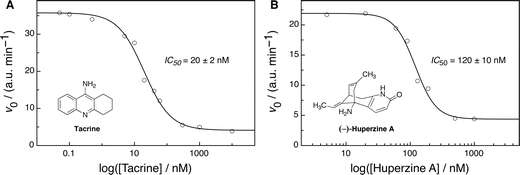 | ||
| Fig. 7 Dose-response curves and associated Hill plot analyses for AChE inhibition by (A) Tacrine and (B) (–)-Huperzine A, respectively, with 0.125 U ml−1 AChE (2.4 nM, determined by UV), 0.5 U ml−1 ChO (60 nM), 40 μM acetylcholine, 700 nM LCG, and 2 μM 1 in 10 mM sodium phosphate buffer at pH 8 (λex = 410 nm, λem = 505 nm). | ||
Determination of absolute acetylcholine and choline concentrations in vitro
Due to the importance of acetylcholine and its precursor/metabolite choline in neurodegenerative diseases, several analytical techniques have been developed for their quantitative detection, including GC, GC-MS, HPLC-ED (i.e., with electrochemical detection), and HPLC-MS.98 Some electrochemical methods, particularly for their detection in food, have also made use of ChO to produce H2O2, measure the total formation of this by-product, and thereby draw conclusions regarding the choline content.78,79 One difficulty of acetylcholine detection is its very low levels in the blood (<20 nM) as well as its very fast hydrolysis by esterases.98 The concentration of choline, its hydrolysis product, lies typically in the micromolar range, e.g., 3–7 μM in canine blood samples.99 In tissues, the levels of acetylcholine and choline can be higher, e.g., 40–80 μM in brain extracts,99,100 reaching a record value of 1 mM acetylcholine in torpedo electric organ tissue,101 from which commercial AChE samples are in fact frequently isolated. In the synaptic cleft of an active neurotransmitter junction the local concentration of acetylcholine is around 100 μM.102 It transpires that a sensitivity in the low μM range is indispensable for the prospective investigation of actual biological samples and tissues.A fluorescence-based supramolecular sensing system would be highly complementary to the established chromatographic procedures since it offers the potential for a real-time monitoring of the concentrations of the analytes. We have established this very application by the enzymatic assays themselves. At a given enzyme concentration, when working below the KM value of the enzyme, the initial reaction rates will increase linearly with the substrate concentration, which provides a direct means of its quantification as an analyte. We have previously utilised enzymatic reactions in combination with supramolecular reporter pairs to quantify different amino acids.40 The enzymatic conversion of the substrates produces a fluorescence response in the presence of the 1·LCG reporter pair, which allows us to set-up calibration lines of the initial rates for determining unknown concentrations of both acetylcholine and choline (Fig. 8); the substrate specificity of the enzymes ensures a highly selective sensing. As can be seen, the method allows accurate determinations (r = 0.998, Fig. 8A) down to the biologically highly relevant low micromolar range.
 | ||
| Fig. 8 (A) Linear relationship between the initial reaction rates and the concentration of choline (5–30 μM with 0.5 U ml−1 ChO, 700 nM LCG, and 2.0 μM 1) and acetylcholine (5–20 μM with 0.1 U ml−1 AChE, 0.5 U ml−1 ChO, 180 nM LCG, and 450 nM 1); in 10 mM sodium phosphate buffer at pH 8 (λex = 410 nm, λem = 505 nm) (B) Domino tandem assay in the presence of both choline and acetylcholine with successive addition of ChO and AChE (0.1 U ml−1 AChE, 0.5 U ml−1 ChO, 180 nM LCG, and 450 nM 1 in 10 mM sodium phosphate buffer at pH 8, λex = 410 nm, λem = 505 nm); the two traces were normalized to the same initial intensity to allow a comparison of the reaction kinetics. | ||
It is even possible to assay both analytes in one and the same sample by means of tandem assays. Note that it would be impossible to differentiate them by a simple displacement assay according to Scheme 1, because both analytes have the same affinity, within error, to calixarene 1 (see also Fig. 6A). To achieve this, we employed a domino tandem assay,14 in which we first added ChO to convert choline, and then, after completion of the first enzymatic reaction, AChE to digest acetylcholine. This results in a stepwise decrease in fluorescence intensity (Fig. 8B), where the rates after addition of the respective enzyme (as well as the intensities of the plateau regions) are again related to the absolute concentrations of each of the two analytes. As can be further seen from the (normalized) traces in Fig. 8B, the initial conversion rate of choline is insensitive to the absolute concentration of acetylcholine. Such determinations are, nevertheless, limited to in vitro experiments, and cannot be transferred to in vivo conditions, for example in the synaptic cleft, where the two analytes are being rapidly interconverted into each other. Thus, while the development of actual biological utilisation is still far-fetched, the presently described integrated enzymological/supramolecular approach has clear-cut advantages in terms of specificity and sensitivity over the previously introduced systems.1–17 It can be transferred beyond simple analyte sensing for biological investigations to the monitoring of enzymatic activity, with the screening of enzyme mutants, activators, and inhibitors being of prime pharmaceutical and biotechnological importance.
Conclusions
The sensing of acetylcholine has remained an ambitious aim in macrocyclic and supramolecular chemistry,1–17 the quest for which has led, among others, to one of the first indicator displacement assays,4 which have later been made popular.18–20 The obstacle of ensuring high selectivity for recognition by the macrocycle, while retaining a sufficient affinity to common fluorescent dyes in order to ease sensing, could not be solved by structural variations and supramolecular design alone. The tandem assays developed herein combine the best out of the 3 worlds of photochemistry, biochemistry, and supramolecular chemistry: We retain fluorescent dyes to ensure sensitive and convenient signalling, we introduce enzymes to facilitate analyte recognition with unparalleled specificity, and we exploit macrocycles to differentiate the substrate and product of the enzymatic reaction, while complexing the fluorescent dye to allow the reporting of the reaction progress through a rapid dynamic equilibrium. All components are integral.The reporter pairs composed of calixarenes 1–3 and LCG operate in water over a broad pH range, show an exceptional fluorescence response (factor of 140), and are tightly bound (up to 107 M−1, depending on ionic strength). In combination with the enzymes acetylcholinesterase and/or choline oxidase it is possible to sense acetylcholine and choline with low micromolar sensitivity, both separately and together. Moreover, these highly sensitive reporter pairs can be used to follow the activity of both enzymes, as well as other enzymes such as amino acid decarboxylases. This enables inhibitor and activator screening by the supramolecular tandem assay methodology, the usefulness of which we have augmented herein by an enzyme-coupled variant, and verified by the observed inhibition exhibited by Alzheimer's drugs.
Acknowledgements
The Chinese team gratefully acknowledges support by the 973 Program (2011CB932502) and NNSFC (20932004). The German team thanks the Deutsche Forschungsgemeinschaft (DFG grant NA-686/5-1), the COST Action CM1005 “Supramolecular Chemistry in Water”, and the Fonds der Chemischen Industrie for support. Both teams thank the Robert Bosch Foundation for bilateral collaborative project support (Wissenschaftsbrücke China). The authors would like to thank Ms. Garima Ghale for advice with enzyme kinetics and Ms. Anja Müller for help with the HPLC-MS measurements.References
- H. J. Schneider, D. Güttes and U. Schneider, Angew. Chem., Int. Ed. Engl., 1986, 25, 647–649 CrossRef.
- H. J. Schneider, D. Güttes and U. Schneider, J. Am. Chem. Soc., 1988, 110, 6449–6454 CrossRef CAS.
- F. Perret, J. P. Morel and N. Morel-Desrosiers, Supramol. Chem., 2003, 15, 199–206 CrossRef CAS.
- M. Inouye, K. Hashimoto and K. Isagawa, J. Am. Chem. Soc., 1994, 116, 5517–5518 CrossRef CAS.
- K. N. Koh, K. Araki, A. Ikeda, H. Otsuka and S. Shinkai, J. Am. Chem. Soc., 1996, 118, 755–758 CrossRef CAS.
- Y. J. Zhang, W. X. Cao and J. Xu, Chin. J. Chem., 2002, 20, 322–326 CAS.
- J. M. Lehn, R. Meric, J. P. Vigneron, M. Cesario, J. Guilhem, C. Pascard, Z. Asfari and J. Vicens, Supramol. Chem., 1995, 5, 97–103 CrossRef CAS.
- H. J. Buschmann, L. Mutihac and E. Schollmeyer, J. Inclusion Phenom. Macrocyclic Chem., 2003, 46, 133–137 CrossRef CAS.
- T. Jin, F. Fujii and Y. Ooi, Sensors, 2008, 8, 6777–6790 Search PubMed.
- T. Jin, Sensors, 2010, 10, 2438–2449 CrossRef CAS.
- T. Jin, J. Inclusion Phenom. Macrocyclic Chem., 2003, 45, 195–201 CrossRef CAS.
- H. Bakirci and W. M. Nau, Adv. Funct. Mater., 2006, 16, 237–242 CrossRef CAS.
- A. Hennig, H. Bakirci and W. M. Nau, Nat. Methods, 2007, 4, 629–632 CrossRef CAS.
- W. M. Nau, G. Ghale, A. Hennig, H. Bakirci and D. M. Bailey, J. Am. Chem. Soc., 2009, 131, 11558–11570 CrossRef CAS.
- H. Bakirci, A. L. Koner, M. H. Dickman, U. Kortz and W. M. Nau, Angew. Chem., Int. Ed., 2006, 45, 7400–7404 CrossRef CAS.
- S. D. Tan, W. H. Chen, A. Satake, B. Wang, Z. L. Xu and Y. Kobuke, Org. Biomol. Chem., 2004, 2, 2719–2721 RSC.
- N. Korbakov, P. Timmerman, N. Lidich, B. Urbach, A. Sa'ar and S. Yitzchaik, Langmuir, 2008, 24, 2580–2587 CrossRef CAS.
- M. Kitamura, S. H. Shabbir and E. V. Anslyn, J. Org. Chem., 2009, 74, 4479–4489 CrossRef CAS.
- A. P. Umali and E. V. Anslyn, Curr. Opin. Chem. Biol., 2010, 14, 685–692 Search PubMed.
- B. T. Nguyen and E. V. Anslyn, Coord. Chem. Rev., 2006, 250, 3118–3127 CrossRef CAS.
- R. Maskiewicz, D. Sogah and T. C. Bruice, J. Am. Chem. Soc., 1979, 101, 5347–5354 Search PubMed.
- R. Maskiewicz, D. Sogah and T. C. Bruice, J. Am. Chem. Soc., 1979, 101, 5355–5364 CrossRef CAS.
- H. Gyllenhammar, J. Immunol. Methods, 1987, 97, 209–213 CrossRef CAS.
- G. Arena, A. Casnati, A. Contino, F. G. Gulino, D. Sciotto and R. Ungaro, J. Chem. Soc., Perkin Trans. 2, 2000, 419–423 RSC.
- C. Bonal, Y. Israeli, J. P. Morel and N. Morel-Desrosiers, J. Chem. Soc., Perkin Trans. 2, 2001, 1075–1078 RSC.
- B. Mokhtari, K. Pourabdollah and N. Dalali, J. Inclusion Phenom. Macrocyclic Chem., 2011, 69, 1–55 Search PubMed.
- G. Arena, A. Contino, F. G. Gulino, A. Magrì, F. Sansone, D. Sciotto and R. Ungaro, Tetrahedron Lett., 1999, 40, 1597–1600 CrossRef CAS.
- N. Douteau-Guével, A. W. Coleman, J. P. Morel and N. Morel-Desrosiers, J. Chem. Soc., Perkin Trans. 2, 1999, 629–633 RSC.
- O. I. Kalchenko, F. Perret, N. Morel-Desrosiers and A. W. Coleman, J. Chem. Soc., Perkin Trans. 2, 2001, 258–263 RSC.
- N. Douteau-Guével, F. Perret, A. W. Coleman, J. P. Morel and N. Morel-Desrosiers, J. Chem. Soc., Perkin Trans. 2, 2002, 524–532 RSC.
- A. Mendes, C. Bonal, N. Morel-Desrosiers, J. P. Morel and P. Malfreyt, J. Phys. Chem. B, 2002, 106, 4516–4524 CrossRef CAS.
- H. Bakirci, A. L. Koner and W. M. Nau, Chem. Commun., 2005, 5411–5413 RSC.
- J. P. Morel and N. Morel-Desrosiers, Org. Biomol. Chem., 2006, 4, 462–465 RSC.
- D. S. Guo, K. Wang and Y. Liu, J. Inclusion Phenom. Macrocyclic Chem., 2008, 62, 1–21 CrossRef CAS.
- N. Basilio, L. García-Río and M. Martín-Pastor, J. Phys. Chem. B, 2010, 114, 7201–7206 Search PubMed.
- H. Bakirci, A. L. Koner and W. M. Nau, J. Org. Chem., 2005, 70, 9960–9966 CrossRef CAS.
- P. Ballester, A. Shivanyuk, A. R. Far and J. Rebek, J. Am. Chem. Soc., 2002, 124, 14014–14016 CrossRef CAS.
- F. Hof, L. Trembleau, E. C. Ullrich and J. Rebek, Angew. Chem., Int. Ed., 2003, 42, 3150–3153 CrossRef.
- R. Ludwig and N. T. K. Dzung, Sensors, 2002, 2, 397–416 CrossRef.
- D. M. Bailey, A. Hennig, V. D. Uzunova and W. M. Nau, Chem.–Eur. J., 2008, 14, 6069–6077 CrossRef CAS.
- A. Praetorius, D. M. Bailey, T. Schwarzlose and W. M. Nau, Org. Lett., 2008, 10, 4089–4092 CrossRef CAS.
- (a) M. Florea and W. M. Nau, Org. Biomol. Chem., 2010, 8, 1033–1039 RSC; (b) G. Ghale, V. Ramalingam, A. R. Urbach and W. M. Nau, J. Am. Chem. Soc., 2011, 133, 7528–7535 CrossRef CAS.
- S. J. Shinkai, K. Araki, T. Matsuda and O. Manabe, Bull. Chem. Soc. Jpn., 1989, 62, 3856–3862 CAS.
- G. Arena, S. Gentile, F. G. Gulino, D. Sciotto and C. Sgarlata, Tetrahedron Lett., 2004, 45, 7091–7094 CrossRef CAS.
- Y. Liu, D. S. Guo, H. Y. Zhang, Y. H. Ma and E. C. Yang, J. Phys. Chem. B, 2006, 110, 3428–3434 CrossRef CAS.
- D. S. Guo, L. H. Wang and Y. Liu, J. Org. Chem., 2007, 72, 7775–7778 CrossRef CAS.
- Y. Liu, D. S. Guo, E. C. Yang, H. Y. Zhang and Y. L. Zhao, Eur. J. Org. Chem., 2005, 162–170 CrossRef CAS.
- Y. Liu, E. C. Yang, Y. Chen, D. S. Guo and F. Ding, Eur. J. Org. Chem., 2005, 4581–4588.
- S. Shinkai, K. Araki, T. Matsuda, N. Nishiyama, H. Ikeda, I. Takasu and M. Iwamoto, J. Am. Chem. Soc., 1990, 112, 9053–9058 CrossRef.
- M. Nishida, D. Ishii, I. Yoshida and S. Shinkai, Bull. Chem. Soc. Jpn., 1997, 70, 2131–2140 CAS.
- The pKa values for deprotonation of the phenoxyl groups are as follows: 3.1 and 12.0 for 1, 4.3, 7.6, and 11.0 for 2, and 2.2, 8.5, 11.6, and 12.0 for 3, cf. I. Yoshida, N. Yamamoto, F. Sagara, D. Ishii, K. Ueno and S. Shinkai, Bull. Chem. Soc. Jpn., 1992, 65, 1012–1015 Search PubMed; H. Matsumiya, Y. Terazono, N. Iki and S. Miyano, J. Chem. Soc., Perkin Trans. 2, 2002, 1166–1172 CAS; J. W. Steed, C. P. Johnson, C. L. Barnes, R. K. Juneja, J. L. Atwood, S. Reilly, R. L. Hollis, P. H. Smith and D. L. Clark, J. Am. Chem. Soc., 1995, 117, 11426–11433 RSC.
- J. Cui, V. D. Uzunova, D. S. Guo, K. Wang, W. M. Nau and Y. Liu, Eur. J. Org. Chem., 2010, 1704–1710 Search PubMed.
- M. Coruzzi, G. D. Andreetti, V. Bocchi, A. Pochini and R. Ungaro, J. Chem. Soc., Perkin Trans. 2, 1982, 1133–1138 RSC.
- A good rule for a convenient and sensitive indicator displacement assay is that the affinity product (defined here as binding constant times concentration) should be about equal for the dye and the analyte. If the binding constant of the dye is 2 orders of magnitude higher than that of the target analyte (e.g., LCG versus choline toward receptor 1), the dye (and host) can be applied in a 2 orders of magnitude lower concentration than that of the analyte, which is particularly desirable for enzymatic reactions in order to minimise potential interferences of the additives.
- Lucigenin fluorescence is sensitive to dynamic quenching by several anions (cf. K. D. Legg and D. M. Hercules, J. Phys. Chem., 1970, 74, 2114–2118 Search PubMed ) which requires attention when selecting the type of buffer and its concentration. Only a few anions, including the perchlorate one selected for the salt-dependent studies (Table 1), show negligible quenching of LCG fluorescence. To avoid the dynamic quenching of LCG by the analyte, micromolar concentrations are preferable for detection and use in enzyme assays.
- C. Huber, K. Fähnrich, C. Krause and T. Werner, J. Photochem. Photobiol., A, 1999, 128, 111–120 CrossRef CAS.
- A. Ikeda and S. Shinkai, Tetrahedron Lett., 1992, 33, 7385–7388 CrossRef CAS.
- R. Ungaro, A. Casnati, F. Ugozzoli, A. Pochini, J. F. Dozol, C. Hill and H. Rouquette, Angew. Chem., Int. Ed. Engl., 1994, 33, 1506–1509 CrossRef.
- A. Ikeda and S. Shinkai, Chem. Rev., 1997, 97, 1713–1734 CrossRef CAS.
- Y. Liu, B. H. Han and Y. T. Chen, J. Phys. Chem. B, 2002, 106, 4678–4687 CrossRef CAS.
- D. Rehm and A. Weller, Isr. J. Chem., 1970, 8, 259–271 CAS.
- G. Diao and Z. Wei, J. Electroanal. Chem., 2004, 567, 325–330 CrossRef CAS.
- I. Spasojević, S. I. Liochev and I. Fridovich, Arch. Biochem. Biophys., 2000, 373, 447–450 Search PubMed.
- K. Wang, D. S. Guo, H. Q. Zhang, D. Li, X. L. Zheng and Y. Liu, J. Med. Chem., 2009, 52, 6402–6412 CrossRef CAS.
- F. Sansone, L. Baldini, A. Casnati and R. Ungaro, New J. Chem., 2010, 34, 2715–2728 RSC.
- F. Perret, A. N. Lazar and A. W. Coleman, Chem. Commun., 2006, 2425–2438 RSC.
- V. D. Uzunova, C. Cullinane, K. Brix, W. M. Nau and A. I. Day, Org. Biomol. Chem., 2010, 8, 2037–2042 RSC.
- T. Loftsson and M. E. Brewster, J. Pharm. Sci., 1996, 85, 1017–1025 CrossRef CAS.
- V. J. Stella and R. A. Rajewski, Pharm. Res., 1997, 14, 556–567 CrossRef CAS.
- B. A. Peddie, M. Lever, C. M. Hayman, K. Randall and S. T. Chambers, FEMS Microbiol. Lett., 1994, 120, 125–131 Search PubMed.
- J. Boch, B. Kempf, R. Schmid and E. Bremer, J. Bacteriol., 1996, 178, 5121–5129 Search PubMed.
- A. Sakamoto, R. Valverde, Alia, T. H. H. Chen and N. Murata, Plant J., 2000, 22, 449–453 Search PubMed.
- D. Ribitsch, W. Karl, E. Wehrschutz-Sigl, S. Tutz, P. Remler, H. J. Weber, K. Gruber, R. Stehr, C. Bessler, N. Hoven, K. Sauter, K. H. Maurer and H. Schwab, Appl. Microbiol. Biotechnol., 2009, 81, 875–886 Search PubMed.
- G. L. Ellman, K. D. Courtney, V. Andres and R. M. Featherstone, Biochem. Pharmacol., 1961, 7, 88–95 CrossRef CAS.
- S. Ikuta, K. Matuura, S. Imamura, H. Misaki and Y. Horiuti, J. Biochem., 1977, 82, 157–163 Search PubMed.
- M. Ohta-Fukuyama, Y. Miyake, S. Emi and T. Yamano, J. Biochem., 1980, 88, 197–203 CAS.
- G. Gadda, Biochim. Biophys. Acta, Proteins Proteomics, 2003, 1646, 112–118 Search PubMed.
- A. Guerrieri and F. Palmisano, Anal. Chem., 2001, 73, 2875–2882 CrossRef CAS.
- G. Panfili, P. Manzi, D. Compagnone, L. Scarciglia and G. Palleschi, J. Agric. Food Chem., 2000, 48, 3403–3407 CrossRef CAS.
- R. C. Wong, T. T. Ngo and H. M. Lenhoff, Int. J. Biochem., 1981, 13, 159–163 Search PubMed.
- A. Hekmat, A. A. Saboury, A. A. Moosavi-Movahedi, H. Ghourchian and F. Ahmad, Acta Biochim. Pol., 2008, 55, 549–557 Search PubMed.
- D. Ribitsch, W. Karl, E. Wehrschütz-Sigl, S. Tutz, P. Remler, H. J. Weber, K. Gruber, R. Stehr, C. Bessler, N. Hoven, K. Sauter, K. H. Maurer and H. Schwab, Appl. Microbiol. Biotechnol., 2009, 81, 875–886 Search PubMed.
- M. Recanatini and P. Valenti, Curr. Pharm. Des., 2004, 10, 3157–3166 Search PubMed.
- E. K. Perry, B. E. Tomlinson, G. Blessed, K. Bergmann, P. H. Gibson and R. H. Perry, Br. Med. J., 1978, 2, 1457–1459 CrossRef CAS.
- R. T. Bartus, Exp. Neurol., 2000, 163, 495–529 Search PubMed.
- S. M. Z. Hossain, R. E. Luckham, A. M. Smith, J. M. Lebert, L. M. Davies, R. H. Pelton, C. D. M. Filipe and J. D. Brennan, Anal. Chem., 2009, 81, 5474–5483 CrossRef.
- M. Israël and B. Lesbats, J. Neurochem., 1981, 37, 1475–1483 Search PubMed.
- S. E. Snyder, N. Gunupudi, P. S. Sherman, E. R. Butch, M. B. Skaddan, M. R. Kilbourn, R. A. Koeppe and D. E. Kuhl, J. Cereb. Blood Flow Metab., 2001, 21, 132–143 Search PubMed.
- A. Guerrieri, G. E. De Benedetto, F. Palmisano and P. G. Zambonin, Analyst, 1995, 120, 2731–2736 RSC.
- For choline oxidase, the excitation wavelength was changed to 410 nm in order to minimise absorption by the flavoprotein in this enzyme, whose characteristic absorption band maxima are around 360 and 455 nm.
- R. J. Kitz and S. Ginsburg, Biochem. Pharmacol., 1968, 17, 525–532 Search PubMed.
- J. Bajgar, P. Hajek, J. Karasova, D. Slizova, O. Krs, K. Kuca, D. Jun, J. Fusek and L. Capek, Int. J. Mol. Sci., 2007, 8, 1165–1176 Search PubMed.
- The equation Ki = (IC50 − 0.5 × [E]0)/(1 + ([S]/KM) was used for conditions of tight stoichiometric competitive inhibition for a monosubstrate reaction, with IC50 determined from the dose-response curves in Fig. 7 and KM = 70 μM for acetylcholine (this work, see text), cf. R. A. Copeland, D. Lombardo, J. Giannaras and C. P. Decicco, Bioorg. Med. Chem. Lett., 1995, 5, 1947–1952 Search PubMed.
- M. Ahmed, J. B. T. Rocha, M. Corrêa, C. M. Mazzanti, R. F. Zanin, A. L. B. Morsch, V. M. Morsch and M. R. C. Schetinger, Chem.-Biol. Interact., 2006, 162, 165–171 Search PubMed.
- X. C. Tang and Y. F. Han, CNS Drug Rev., 1999, 5, 281–300 Search PubMed.
- C. Costagli and A. Galli, Biochem. Pharmacol., 1998, 55, 1733–1737 CrossRef CAS.
- S. Darvesh, R. Walsh and E. Martin, Cell. Mol. Neurobiol., 2003, 23, 93–100 Search PubMed.
- T. H. Tsai, J. Chromatogr., Biomed. Appl., 2000, 747, 111–122 CrossRef CAS.
- A. K. Singh and L. R. Drewes, J. Chromatogr., Biomed. Appl., 1985, 339, 170–174 Search PubMed.
- M. H. Aprison and P. Nathan, Arch. Biochem. Biophys., 1957, 66, 388–395 Search PubMed.
- M. Israël and B. Lesbats, Neurochem. Int., 1981, 3, 81–90 Search PubMed.
- B. Alberts, A. Johnson, J. Lewis, M. Raff, K. Roberts and P. Walter, in Molecular Biology of the Cell, Gerland Publishing, New York, 5th edn, 2008, ch. 15, p. 882 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Expanded introduction, experimental section, and additional results, including all direct host–guest titrations and Job's plot analyses, calorimetric titrations, NMR measurements, ORTEP view of XRD structure, fluorescence lifetime measurements, cyclic voltammetry data, direct host–guest titrations in the enzymatic buffers, supramolecular tandem assay results for lysine decarboxylase, Lineweaver–Burk plots, mass spectrometry measurements, and fluorescence control experiments for the enzyme assays. CCDC reference number 820542. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00231g |
| This journal is © The Royal Society of Chemistry 2011 |