Thermotropic liquid crystals based on 1,8,9,16-tetrasubstituted tetraphenylenes and their structure–property relationship studies†
Chun-Kit
Hau
abc,
Stephen Sin-Yin
Chui
d,
Wei
Lu
d,
Chi-Ming
Che
cde,
Ping-Shing
Cheng
ab,
Thomas C. W.
Mak
ab,
Qian
Miao
ab and
Henry N. C.
Wong
*abce
aDepartment of Chemistry, The Chinese University of Hong Kong, Shatin, New Territories, Hong Kong SAR, China
bCentre of Novel Functional Molecules, The Chinese University of Hong Kong, Shatin, New Territories, Hong Kong SAR, China
cInstitute of Molecular Functional Materials, An Area of Excellence Scheme established under the University Grants Committee, Hong Kong
dDepartment of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong SAR, China
eState Key Laboratory on Synthetic Chemistry, Pokfulam Road and Shatin, Hong Kong SAR, China
First published on 28th March 2011
Abstract
Twelve 1,8,9,16-tetraalkoxytetraphenylenes were synthesized and characterized, five of which exhibiting thermotropic liquid crystal properties were examined by differential scanning calorimetry (DSC), polarizing-light optical microscopy (POM) and variable-temperature X-ray diffractometry (VTXRD). The structure–property relationship of each mesomorphic solid is discussed in the context of the racemic/non-racemic nature of the sample, different chain length of n-alkyl substituents, effect of chain lipophilicity and intermolecular interactions.
Introduction
Tetraphenylene (tetrabenzo[a,c,e,g]cyclooctatetraene) is a structurally unique molecule belonging to a D2d symmetry point group.1 The four benzene rings alternate above and below the mean plane of the molecule, resulting in a rigid saddle conformation.2,3 Due to the high inversion barrier of the central cyclooctatetraene,4 a chiral version of tetraphenylenes can be obtained by introducing substituents at appropriate sites.Over the past decades, a number of non-planar molecules have been shown to exhibit interesting liquid crystal properties.5–12 Based on the extensive studies in the past, tetraphenylenes can be regarded as a promising candidate of a new structural core for liquid crystal materials. For example, Laschat and co-workers reported tetraphenylenes with eight peripheral chains (i.e.alkoxy, alkanoate, p-alkoxybenzoate and gallic ester) to exhibit thermotropic columnar and smectic mesophases.13 Furthermore, such discotic tetraphenylenes were discovered to have anomalous odd–even effects, with respect to the change of transition temperatures in relation to the number of methylene groups in the carbon side chains.13c, d
To the best of our knowledge, related studies on the liquid crystal properties of chiral tetraphenylenes have hitherto been unexplored. Herein, we disclose the synthesis of a group of tetraalkoxy-substituted tetraphenylenes in both non-racemic and racemic forms, and the results of the studies on their liquid crystal properties by polarizing-light optical microscopy (POM) and on the phase transitions associated with the mesogenic members by differential scanning calorimetry (DSC) and variable-temperature X-ray diffractometry (VTXRD). Among those mesomorphic derivatives, compound (±)-11 is taken as an example for an in-depth discussion because of its significant structural change based on its single-crystal X-ray structure and subsequent VTXRD studies.
Results and discussion
Synthesis and characterization
To study the liquid crystal properties of the tetraalkoxytetraphenylenes derived from tetraphenylenol 1,14 two different series of derivatives were designed and synthesized.As illustrated in Scheme 1, tetraphenylenol 1 was first treated with (S)-camphorsulfonyl chloride to give the two diastereoisomers of tetrakis-(S)-camphorsulfonates (R,R)-2a and (S,S)-2b, which were separated by column chromatography. Upon hydrolysis, (R,R)-1 was obtained in good yield from (R,R)-2a.
![Reagents and conditions: (i) R*Cl, Et3N, THF, 0–23 °C, (R,R)-2a: 45%; (S,S)-2b: 47%. (ii) KOH (aq.), MeOH, reflux, 97%. (iii) RX, Cs2CO3, DMF, 80 °C [(R,R)-3: RX = C4H9Cl; 4: RX = C7H15Br; 5: RX = C12H25Br; 6: RX = TsO(C2H4)2CH3; (±)-7: RX = TsO(C2H4)4CH3]](/image/article/2011/SC/c1sc00053e/c1sc00053e-s1.gif) | ||
| Scheme 1 Reagents and conditions: (i) R*Cl, Et3N, THF, 0–23 °C, (R,R)-2a: 45%; (S,S)-2b: 47%. (ii) KOH (aq.), MeOH, reflux, 97%. (iii) RX, Cs2CO3, DMF, 80 °C [(R,R)-3: RX = C4H9Cl; 4: RX = C7H15Br; 5: RX = C12H25Br; 6: RX = TsO(C2H4)2CH3; (±)-7: RX = TsO(C2H4)4CH3] | ||
As a measure to impart liquid crystalline properties in the resulting systems, the phenolic groups in (R,R)-1 were alkylated with C4-, C7-, C12-, and di(ethylene glycol) methyl ether-derived side chains to provide (R,R)-3–6. Similarly, racemic (±)-4–7 were prepared.
In addition, two sets of side chains having different lipophilicities could be readily anchored onto the tetrahydroxytetraphenylene core by the route shown in Scheme 2. The sequence involved in the following steps: 1: protection of the proximal pair of hydroxyls with 1,2-bis(bromomethyl)benzene, 2: alkylation of the free hydroxyls with an alkyl halide, 3: deprotection by hydrogenolysis, and 4: further alkylation with a polyoxy alkyl tosylate. In this manner, (S,S)-11,12 and (±)-11,12 were obtained.
![Reagents and conditions: (i) KOH (aq.), MeOH, reflux, 97%. (ii) Cs2CO3, DMSO, 80 °C, 38%. (iii) RX, Cs2CO3, DMF, 80 °C [(S,S)-9: RX = C7H15Br; (S,S)-10: RX = C12H25Br]; H2, Pd/C, THF; (iv) Cs2CO3, DMF, 80 °C [(S,S)-11: RX = TsO(CH2CH2O)2CH3; (S,S)-12: RX = TsO(CH2CH2O)4CH3]](/image/article/2011/SC/c1sc00053e/c1sc00053e-s2.gif) | ||
| Scheme 2 Reagents and conditions: (i) KOH (aq.), MeOH, reflux, 97%. (ii) Cs2CO3, DMSO, 80 °C, 38%. (iii) RX, Cs2CO3, DMF, 80 °C [(S,S)-9: RX = C7H15Br; (S,S)-10: RX = C12H25Br]; H2, Pd/C, THF; (iv) Cs2CO3, DMF, 80 °C [(S,S)-11: RX = TsO(CH2CH2O)2CH3; (S,S)-12: RX = TsO(CH2CH2O)4CH3] | ||
It was hoped that the twelve enantiopure and racemic long chain tetraalkoxytetraphenylenes in hand would allow us to unveil the relationship between their liquid crystalline properties and the nature and length of the appended side chains, and, in particular, the racemic or non-racemic nature of the samples.
Liquid crystal properties
The liquid crystal properties of the synthesized tetraphenylene derivatives were investigated by differential scanning calorimetry (DSC), polarizing-light optical microscopy (POM) and in situ variable temperature X-ray diffraction (VTXRD). The transition temperatures and enthalpy changes of each sample observed in DSC study are listed in Table 1.| Compound | Mesophase | Phase transitionsb | T/°C (ΔH/J g−1) |
|---|---|---|---|
| a Two times heating–cooling cycle were done for each sample and the data listed was recorded at 2nd heating–cooling cycle. b Cr = crystal phase, I = isotropic liquid. | |||
| (R,R)-3 | N | Cr → I; I → Cr | 133.8 (5.6); 112.1 (−10.0) |
| (R,R)-4 | Y | Cr → SmA; SmA → I; I → SmA; SmA → Cr | 27.7 (1.0); 116.5 (2.8); 89.4 (−4.0); 23.0 (−1.1) |
| (R,R)-4 (50% ee) | Y | Cr → SmA; SmA → I; I → SmA; SmA → Cr | 31.7 (7.5); 138.9 (29.2); 111.8 (−31.1); 24.1 (−8.0) |
| (±)-4 | Y | Cr → SmA; SmA → I; I → SmA; SmA → Cr | 29.3 (13.4); 141.5 (61.2); 110.7 (−63.4); 23.4 (−14.1) |
| (R,R)-5 | N | Cr1 → Cr2; Cr2 → I; I → Cr2; Cr2 → Cr1 | 48.8 (2.9); 101.6 (9.5); 81.3 (−10.0); 51.9 (−3.3) |
| (±)-5 | N | Cr1 → Cr2; Cr2 → Cr3 → I; I → Cr2; Cr2 → Cr1 | 54.5 (11.0); 104.1 (28.8); 91.2 (−32.6); 52.1 (−12.2) |
| (R,R)-6 | N | — | — |
| (±)-6 | N | Cr1 → Cr2; Cr2 → I; I → Cr1 | 76.1 (3.7); 91.4 (28.7); 64.5 (−35.6) |
| (±)-7 | N | Cr → I; I → Cr | 31.3 (23.6); 18.2 (−19.2) |
| (S,S)-11 | N | — | — |
| (±)-11 | Y | Cr1 → SmA; SmA → I; I → SmA; SmA → Cr2 | 25.1 (4.2); 125.3 (57.9); 103.8 (−59.2); 25.1 (−5.3) |
| (S,S)-12 | N | — | — |
| (±)-12 | Y | Cr → Sm; Sm → I; I → Sm; Sm → Cr | 44.1 (1.6); 84.6 (7.8); 76.2 (−8.7); 48.4 (−1.6) |
The results of DSC studies are listed in Table 1, the nature of mesophases of (R,R)-4, (R,R)-4 (50% ee), (±)-4, (±)-11 and (±)-12 were identified according to the classification system described by Kumar and Dierking.15 As depicted in Fig. 1–3, (R,R)-4, (R,R)-4 (50% ee) and (±)-4 displayed enantiotropic smectic A mesophases, as confirmed by the characteristic fan-shaped texture observed in POM images upon cooling the corresponding isotropic liquids. However, both (R,R)-4 (50% ee) and (±)-4 show a more extended fan-shaped texture than that of (R,R)-4. Similarly, (±)-11 was found to display an enantiotropic smectic A mesophase according to the typical fan-shaped texture (Fig. 4). (±)-12 displayed an enantiotropic highly ordered smectic mesophase, as confirmed by the characteristic broken-fan shaped texture upon cooling of the isotropic liquid (Fig. 5).
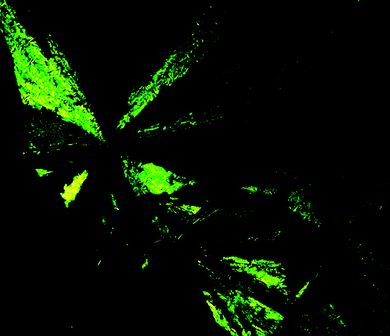 | ||
| Fig. 1 Optical photomicrographs (magnification 50×) showing the fan-shaped texture of the smectic A mesophase of compound (R,R)-4 at 60 °C on cooling. | ||
 | ||
| Fig. 2 Optical photomicrographs (magnification 50×) showing the fan-shaped texture of the smectic A mesophase of compound (R,R)-4 (50% ee) at 60 °C on cooling. | ||
 | ||
| Fig. 3 Optical photomicrographs (magnification 50×) showing the fan-shaped texture of the smectic A mesophase of compound (±)-4 at 80 °C on cooling. | ||
 | ||
| Fig. 4 Optical photomicrographs (magnification 50×) showing the fan-shaped texture of the smectic A mesophase of compound (±)-11 at 60 °C on cooling. | ||
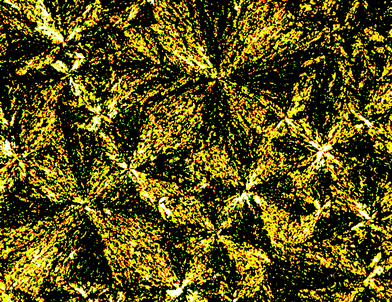 | ||
| Fig. 5 Optical photomicrographs (magnification 50×) showing the broken fan-shaped texture of the highly ordered smectic mesophase of compound (±)-12 at 70 °C on cooling. | ||
According to the DSC results, the formation of the mesophases was found to be affected by the enantiopurity of the compound and the nature of the peripheral alkoxy chains. Both racemic 4 and its optical pure (R,R)-4 were found to be mesomorphic. In both cases, they tend to form the smectic A mesophase, based on POM results (Fig. 1 and 3). Compound (±)-4 was found to have a lower melting point and higher clearing point than the corresponding compound (R,R)-4. In addition, the enthalpy change of (±)-4 was found to be larger than that of (R,R)-4. It is noteworthy that both temperature range and enthalpy changes were significantly increased when the optical purity of (R,R)-4 dropped to 50% ee. Unlike the case in compound 4, compounds 3 and 5 showed no liquid-crystalline behavior, according to the DSC data and POM studies. This indicates that the alternation of the length of peripheral n-alkoxy chains would result in a detrimental effect to the liquid-crystalline properties of the corresponding compounds.
Similarly, neither racemic form nor optically pure form of compounds 6 and 7 were found to show mesogenic properties. This reflects that the peripheral polyether chains are unfavorable for the development of liquid-crystalline properties.
Compounds 11 and 12 possess two lipophilic chains and two lipophobic chains, which show different mesomorphic behavior from that of 4. Both optically pure forms of compounds 11 and 12 show no liquid-crystalline behavior. Substituting two lipophilic chains into polyether chains suppressed the mesophase formation of the racemic forms of compound 11 and 12, in terms of the transition temperature range and enthalpy changes.
The phase transitions of the selected compounds (R,R)-4, (R,R)-4 (50% ee), (±)-4, (±)-11 and (±)-12 were investigated by in situvariable-temperature X-ray diffraction. Fig. 6a shows the X-ray diffractogram of (R,R)-4 recorded upon heating the sample from 30 to 180 °C and cooling the isotropic liquid from 180 to 30 °C. At 80 °C, a Cr → SmA transition occurred, as indicated by the shift of the diffraction peak from its original position (2θ value is 3.73°) to a new position with a 2θ value of 3.60°. This implied that the d-spacing value of the lattice expanded from the original 23.6 to 24.5 Å as the temperature increased. As the temperature increased, the peak became more intense until the temperature reached 180 °C. The peak vanished when (R,R)-4 became an isotropic liquid at 180 °C.
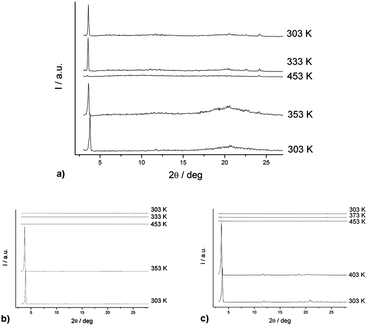 | ||
| Fig. 6 (a) In situ variable-temperature diffractogram of (R,R)-4, (b) in situ variable-temperature diffractogram of (R,R)-4 (50% ee), (c) in situ variable-temperature diffractogram of (±)-4, all are recorded upon heating the sample in 30–180 °C and cooling its isotropic liquid from 180 to 30 °C. | ||
Similar lamellar arrangements of molecules in this mesophase likely exist with a structural resemblance to that in the crystalline structure. During cooling the sample to 60 °C, a new peak with a 2θ value of 3.55° reappeared. As the mesophase was cooled to 30 °C, the peak was further shifted to a position with a 2θ value of 3.58°. This indicated the lattice was showing a tendency to constrict from the mesophase into a more ordered crystalline phase.
Similar observations can also be found in VTXRD analysis of (R,R)-4 (50% ee) and (±)-4, as depicted in Fig. 6b and c. The results are consistent with the DSC and POM studies and the observed single diffraction peaks support the formation of smectic A mesophase with characteristic lamellar distances of 23.5 and 25.2 Å, respectively. However, decrease in intensity of this peak was noted in both cases when the isotropic liquid was cooled. This might be explained by the observation that the isotropic liquid became less viscous and moved away from the sample holder. This is also consistent with its waxy texture of the compounds before the analysis.
The in situ variable-temperature diffractogram of (±)-11 is shown in Fig. 7a. At room temperature, (±)-11 exists as a well-oriented crystalline structure with observed peaks at 7.13 and 7.74°. At 70 °C, a new diffraction peak appeared with a 2θ value of 3.79° and this peak became more intense as the temperature increased. This peak corresponds to a lamellar distance of 23.5 Å of the smectic A mesophase. Upon cooling from its isotropic liquid, this diffraction peak reappeared at a similar position at 90 °C. Moreover, some high-angle weak peaks became more visible and one sharp peak with a 2θ of 20.8° (d = 4.26 Å) was noted. Both peaks also showed a tendency to shift back to their original positions in the crystalline phase. These observations strongly suggest that a similar lamellar arrangement of molecules in the mesophase likely exist, which was completely different from that in the original solid sample.
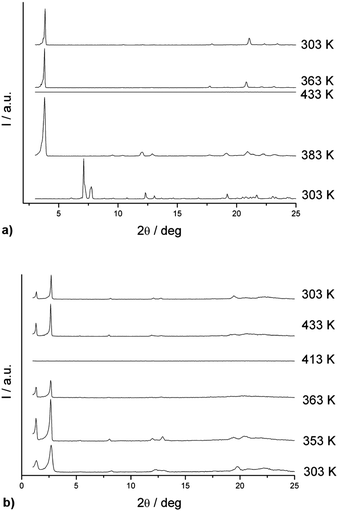 | ||
| Fig. 7 (a) In situ variable-temperature diffractogram of (±)-11 recorded upon heating the sample in 30–160 °C and cooling its isotropic liquid from 160 to 30 °C. (b) In situ variable-temperature diffractogram of (±)-12 recorded upon heating the sample from 30 to 140 °C and cooling its isotropic liquid from 140 to 30 °C. | ||
Similarly, the in situ variable-temperature diffractogram of (±)-12 indicated shifts of peaks as the temperature increased, also the intensities of peaks became more intense at 80 °C. Two diffraction peaks with 2θ values at 1.31° and 2.65° were found, that correspond to interlayer distances of 67.3 and 33.5 Å, respectively, for the [100] and [200] Miller planes of the lamellar highly ordered smectic mesophase. Reappearance of the diffraction peaks in the cooling cycle of (±)-12 reveals that the molecular arrangement in the mesophase is a reversible process and little structural change happened when the isotropic liquid was cooled to its crystal state.
X-Ray crystal structure of (±)-11
For better understanding on the structural change of (±)-11 upon heating, we obtained single crystals and determined its X-ray structure. Single crystals of (±)-11 suitable for X-ray diffraction determination were obtained by slow evaporation of its solution in CH2Cl2 at room temperature. The crystallographic data are summarized in the notes and reference section.‡Fig. 8 shows the crystal structure of (±)-11 and the packing of the molecules in crystalline state. It crystallized in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and as shown in Fig. 8b, the molecules are held by weak π–π stacking interactions (C2–C3 (1 + x, y, 1 + z)⋯C2′–C3′ (1 − x, 1 − y, 1 − z) 3.48 Å and C8′–C9′ (1 − x, 1 − y, 1 − z)⋯C8–C9 (1 + x, y, z) 3.77 Å) between the two opposite enantiomers of (±)-11 (which are labeled in green and yellow color, respectively). Because of these weak interactions, the two different enantiomers of (±)-11 are packed in a zigzag chain along the c-axis.
and as shown in Fig. 8b, the molecules are held by weak π–π stacking interactions (C2–C3 (1 + x, y, 1 + z)⋯C2′–C3′ (1 − x, 1 − y, 1 − z) 3.48 Å and C8′–C9′ (1 − x, 1 − y, 1 − z)⋯C8–C9 (1 + x, y, z) 3.77 Å) between the two opposite enantiomers of (±)-11 (which are labeled in green and yellow color, respectively). Because of these weak interactions, the two different enantiomers of (±)-11 are packed in a zigzag chain along the c-axis.
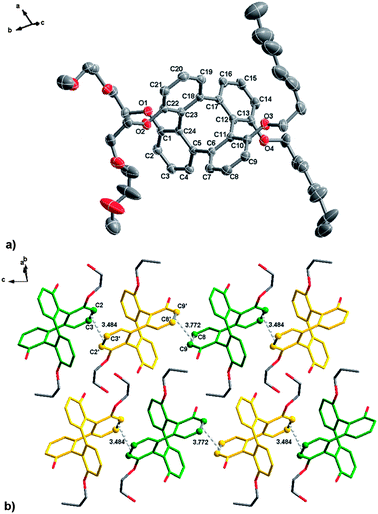 | ||
| Fig. 8 (a) ORTEP diagram of compound (±)-11. Thermal ellipsoids were plotted at 30% probability level. (b) Weak π–π interactions are shown and hydrogen atoms are removed for clarity. | ||
Since there is no solvent molecules be trapped in the crystal lattice in the X-ray structure of (±)-11 it is used as a starting model to explain the structural change when its crystalline solid was heated to the mesophase. As depicted In Fig. 7a, at room temperature, the observed PXRD pattern roughly matched the simulated pattern calculated by the X-ray structure (not shown), regardless of the variation of intensity of peaks due to the effect of preferred orientation of crystallites in the solid. Particularly, the two observed peaks at 7.13 and 7.74° (2θ) matched with the calculated values for the reflections [010] and [100], respectively. Upon heating to 100 °C, such observed XRD pattern completely vanished, and a low-angle strong peak was found, that could be explained by the layered structure of the smectic mesophase. The observed interlayer spacing of 23.1 Å was comparable to the modified molecular shape of (±)-11, where the four chains are stretched out along the same direction. Taking into consideration of the chain lengths of the n-hexyl and polyethoxy substituents and approximate size of the central tetraphenylene core, a molecular arrangement shown in Fig. 9 is proposed in the mesophase, that might account for the observed d-value found in the XRD pattern collected at 100 °C.
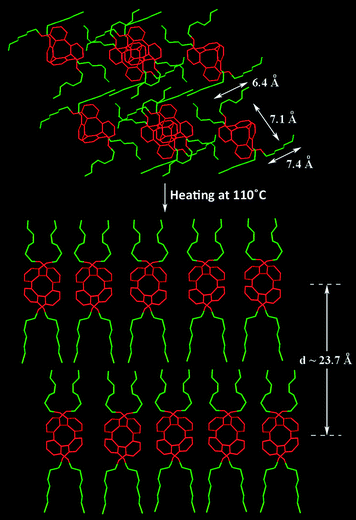 | ||
| Fig. 9 Illustration diagram for the transformation process of (±)-11 from its crystal structure to liquid crystalline mesophase. | ||
Self-assembly properties in polar solvent
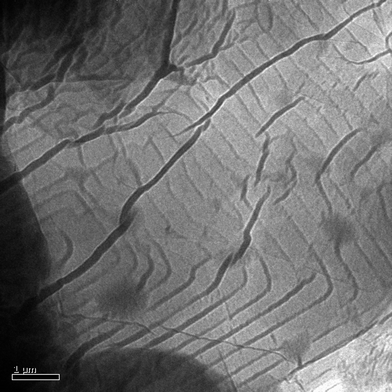
All the compounds that we synthesized are expected to show different lipophilicities in polar or organic solvents. We are also interested in exploring for any possible self-assembly of such compounds, especially amphiphilic compounds such as 11 and 12 in polar solvents, to make supramolecular nanostructures. Compounds 5, 6, 11 and 12 were heated to dissolve them in ethanol–water solutions. Upon cooling of the clear solution, the precipitates obtained were subjected for TEM and SEM analysis. For chiral compounds (R,R)-5 and (R,R)-6, no observable structural textures was observed. This may be due to waxy nature of their solid state at room temperature which hampers the precipitation process.Racemic compound 11 was found to form micro-sized wires (Fig. 10). The primary growth direction of such wires were determined to follow along the [100] axis, which is also consistent with the X-ray result of (±)-11. This implies that the central tetraphenylene core provides a directing role in the formation of such crystalline supramolecular microstructures.
![TEM (A) and its corresponding SAED (B) images recorded with a single microwire of compound (±)-11. The primary growth direction of the microwire is along the [100] axis, as shown in (C). Image (D) is a SAED pattern simulated from the X-ray single-crystal data of compound (±)-11 which matches well to the experimental SAED pattern.](/image/article/2011/SC/c1sc00053e/c1sc00053e-f10.gif) | ||
| Fig. 10 TEM (A) and its corresponding SAED (B) images recorded with a single microwire of compound (±)-11. The primary growth direction of the microwire is along the [100] axis, as shown in (C). Image (D) is a SAED pattern simulated from the X-ray single-crystal data of compound (±)-11 which matches well to the experimental SAED pattern. | ||
Precipitation of (±)-12 from hot ethanol–water solution was found to lead to stacked sheet-like nanostructures, as observed under a transition electron microscope (TEM, Fig. 11). The lateral sizes and thickness of these nanosheets are estimated to be tens of micrometres and less than 100 nm, respectively. This is supportive of the smectic mesophases observed with (±)-12 at elevated temperatures. Such self-assembled lamellar nanostructures are presumably attributed to the amphililic properties of compound (±)-12. A slight increasing at the length of the hydrophilic oligoethylene glycol chains may alter the self-assembling process, as evidenced by the contrast that (±)-11 forms microwires whereas (±)-12 forms nanosheets under identical conditions.
Discussion
In this work, the effect of the racemic and non-racemic nature of central tetraphenylene core, the nature and the chain length of the peripheral alkoxy chains on their mesomorphic properties have been demonstrated. Among (R,R)-3, (R,R)-4 and (R,R)-5, only (R,R)-4 shows mesomorphic behavior. Since all of them possess the same molecular symmetry, the chain length will be the only reason for such mesomorphic behavior difference. According to DSC results, (R,R)-3 was found to be non-mesogenic. This indicates that a suitable length of the peripheral chains against the central tetraphenylene core is necessary. However, when the peripheral chains are too long (12-carbons) in relation to the tetraphenylene core size, it may generate a higher steric congestion, which results in an unfavorable effect on the formation of a more ordered mesophase. The seven-carbon length of n-alkoxy chains was found to be most suitable for the formation of new mesophases.In the case of compound 4, the mesomorphic character was found to increase as the optical purity of the compound decreased, based on DSC and POM results. With reference to the crystal data of (±)-11, a more oriented and effective packing of the tetraphenylenes were found in the racemic mixture. Such kind of packing was believed to be less stable when the central tetraphenylene cores are enantiopure in nature. According to the POM result of (R,R)-4, only a limited amount of fan-shaped structures of smectic A mesophase was found. Also the small enthalpy change for the SmA → I or I → SmA in the case of (R,R)-4 reveals that the long-range positional and orientational orders were the least among the three different optical purity samples, as a result of molecular chirality. The mesomorphic behaviors of (R,R)-4 was further improved after decreasing the enantiopurity to 50% ee, providing further proof for this interpretation.
With reference to the crystal data of (±)-11, the two polyether chains were observed to be more irregularly folded, as compared with the n-heptyloxy chains. Both compounds 6 and 7 did not form any mesophases, and therefore the packing of molecules at elevated temperature was proven to be unstable.
Both enantiopure compounds 11 and 12 did not form any mesophases, and therefore the packing of the molecules with the same chirality was proven to be more interrupted due to the presence of irregular folding polyether chains. By the same reason, the mesomorphic behavior of (±)-11 was less effective than that of (±)-4 in terms of enthalpy change and temperature range as a result of molecular non-symmetry. According to the crystal data of (±)-11, the molecules were found to be tilt arranged in a small extent. Such tilted arrangement of molecules was believed to be further magnified in the case of (±)-12. Consequently, this circumstance facilitates the formation of a highly ordered smectic mesophase.
Conclusions
Twelve 1,8,9,16-tetrahydroxytetraphenylene derivatives were prepared and synthesized. Depending on the length, nature of the peripheral n-alkoxy chains and the racemic and non-racemic nature of the central tetraphenylene core, mesomorphic properties ranging from smectic A to highly ordered smectic mesophases were observed. Variation in the enantiopurity of the tetraphenylene core was found to significantly tune the stability of the smectic mesophase at elevated temperature. Racemic compound 12 was also found to form stacking nanosheets through self-assembly in polar solvent, which is a good starting point for further studies and explorations. Thus the amphiphilic tetraphenylenes could be useful molecular building blocks for supramolecular soft nanomaterials.Acknowledgements
The work described in this project is supported by a grant from the Research Grants Council of the Hong Kong Special Administrative Region, China (RGC ref. No. CUHK403909), as well as partially by the Area of Excellence Scheme established under the University Grants Committee of Hong Kong Special Administrative Region, China (AoE/P-03/08). We thank Dr Tze-Lock Chan and Prof. Sabine Laschat for their helpful comments and suggestions.Notes and references
- For reviews: (a) T. C. W. Mak and H. N. C. Wong in Comprehensive Supramolecular Chemistry, ed. D. D. MacNicol, F. Toda and P. Bishop, Pergamon Press, Oxford, 1996, vol. 6, pp. 351–369 Search PubMed; (b) T. C. W. Mak and H. N. C. Wong, Top. Curr. Chem., 1987, 140, 141–164 CAS; (c) X.-L. Hou, H. Huang and H. N. C. Wong, Synlett, 2005, 1073–1089 CAS; (d) A. Rajca, S. Rajca, M. Pink and M. Miyasaka, Synlett, 2007, 1799–1822 CrossRef CAS; (e) M. J. Marsella, S. Rahbarnia and N. Wilmot, Org. Biomol. Chem., 2007, 5, 391–400 RSC; (f) C. Wang and Z. Xi, Chem. Commun., 2007, 5119–5133 RSC; (g) I. Hisaki, M. Sonoda and Y. Tobe, Eur. J. Org. Chem., 2006, 833–847 CrossRef CAS; (h) H. Huang, C.-K. Hau, C. C. M. Law and H. N. C. Wong, Org. Biomol. Chem., 2009, 7, 1249–1257 RSC.
- I. L. Karle and L. O. Brockway, J. Am. Chem. Soc., 1944, 66, 1974–1979 CrossRef CAS.
- H. Irngartinger and W. R. K. Reibel, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1981, 37, 1724–1728 CrossRef.
- (a) H. Huang, S. Stewart, M. Gutmann, T. Ohhara, N. Niimura, Y.-X. Li, J.-F. Wen, R. Bau and H. N. C. Wong, J. Org. Chem., 2009, 74, 359–369 CrossRef CAS; (b) S. M. Bachrach, J. Org. Chem., 2009, 74, 3609–3611 CrossRef CAS.
- (a) S. Sergeyev, W. Pisula and Y. H. Geerts, Chem. Soc. Rev., 2007, 36, 1902–1929 RSC; (b) S. Laschat, A. Baro, N. Steinke, F. Giesselmann, C. Haegele, G. Scalia, R. Judele, E. Kapatsina, S. Sauer, A. Schreivogel and M. Tosoni, Angew. Chem., Int. Ed., 2007, 46, 4832–4887 CrossRef CAS; (c) J. W. Goodby, in Handbook of Liquid Crystals, Wiley-VCH, Weinheim, 1998, vol. 2A, pp. 411–440 Search PubMed.
- (a) C. Tschierske, Annu. Rep. Prog. Chem., Sect. C, 2001, 97, 191–267 RSC; (b) C. Tschierske, J. Mater. Chem., 1998, 8, 1485–1508 RSC.
- Examples of tribenzocyclononenes, see: (a) Z. Luz, R. Poupko, E. J. Wachtel, H. Zimmermann and V. Bader, Mol. Cryst. Liq. Cryst., 2003, 397, 367–377 CAS; (b) H. Zimmermann, V. Bader, R. Poupko, E. J. Wachtel and Z. Luz, J. Am. Chem. Soc., 2002, 124, 15286–15301 CrossRef CAS; (c) A. Jakli, M. Müller and G. Heppke, Liq. Cryst., 1999, 22, 945–952; (d) H. Budig, R. Lunkwitz, R. Paschke, C. Tschierske, U. Nütz, S. Diele and G. Pelzl, J. Mater. Chem., 1996, 6, 1283–1289 RSC.
- Examples of tetrabenzocyclododecatetraene, see: (a) V. Percec, C. G. Cho and C. Pugh, J. Mater. Chem., 1991, 1, 217–222 RSC; (b) V. Perec, C. G. Cho and C. Pugh, Macromolecules, 1991, 24, 3227–3234 CrossRef CAS.
- Examples of metacyclophanes, see: (a) A. Arcioni, R. Tarroni, C. Zannoni, E. Dalcanale and A. Du Vosel, J. Phys. Chem., 1995, 99, 15981–15986 CrossRef CAS; (b) E. Dalcanale, A. Du Vosel, A. M. Levelut and J. Malthête, Liq. Cryst., 1991, 10, 185–198 CrossRef CAS; (c) G. Cometti, E. Dalcanale, A. Du Vosel and A.-M. Levelut, J. Chem. Soc., Chem. Commun., 1990, 163–165 RSC.
- Examples of calix[4]arenes, see: (a) B. Xu and T. M. Swager, J. Am. Chem. Soc., 1993, 115, 1159–1160 CrossRef CAS; (b) T. Komori and S. Shinkai, Chem. Lett., 1993, 1455–1458 CAS.
- Example of self-assembly of dendrimers, see: V. Percec, M. Glodde, G. Johansson, V. S. K. Balagursamy and P. A. Heiney, Angew. Chem., Int. Ed., 2003, 42, 4338–4342 Search PubMed.
- Examples of triazacyclononanes, see: (a) S. Schmidt, G. Lattermann, R. Kleppinger and J. H. Wendorff, Liq. Cryst., 1994, 16, 693–702 CAS; (b) J.-M. Lehn, J. Malthête and A.-M. Levelut, J. Chem. Soc., Chem. Commun., 1985, 1794–1796 RSC.
- (a) E. Wuckert, M. Dix, S. Laschat, A. Baro, J. L. Schulte, C. Hägele and F. Giesselmann, Liq. Cryst., 2004, 31, 1305–1309 CrossRef CAS; (b) E. Wuckert, S. Laschat, A. Baro, C. Hägele, F. Giesselmann and H. Luftmann, Liq. Cryst., 2006, 33, 103–107 CrossRef CAS; (c) C. Hägele, E. Wuckert, S. Laschat and F. Giesselmann, ChemPhysChem, 2009, 10, 1291–1298 CrossRef; (d) E. Wuckert, C. Hägele, F. Giesselmann, A. Baro and S. Laschat, Beilstein J. Org. Chem., 2009, 5 Search PubMed.
- H.-Y. Peng, C.-K. Lam, T. C. W. Mak, Z. Cai, W.-T. Ma, Y.-X. Li and H. N. C. Wong, J. Am. Chem. Soc., 2005, 127, 9603–9611 CrossRef CAS.
- (a) D. Demus and L. Richter, Textures of Liquid Crystals, Verlag, Chemie, Weinheim, 1984 Search PubMed; (b) I. Dierking Textures of Liquid Crystals, Wiley-VCH, Weinheim, 2003 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC reference number 810216. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00053e |
| ‡ Crystal data of (±)-11: C48H64O8, Mr = 769.0, triclinic, P, a = 12.6286(9), b = 13.2786(10), c = 14.6898(10) Å, α = 85.812(2), β = 77.077(2), γ = 67.470(2)°, V = 2217.5(3) Å3, Z = 2, T = 296 K, μMo = 0.08 mm−1, F(000) = 832, total number of observed data [F2 > 2σI] = 42408 and independent reflections 8687 (Rint = 0.097). Number of parameters = 505, goodness of fit = 1.002, R1 = 0.067 and wR2 = 0.2093. |
| This journal is © The Royal Society of Chemistry 2011 |