Transition metal-catalyzed cross coupling with N-acyliminium ions derived from quinolines and isoquinolines†
Thomas J. A.
Graham
,
Jason D.
Shields
and
Abigail G.
Doyle
*
Department of Chemistry, Princeton University, Washington Rd, Princeton, New Jersey 08544, USA. E-mail: agdoyle@princeton.edu; Fax: +1 609 258 2558; Tel: +1 609 258 3937
First published on 21st February 2011
Abstract
A Ni(0) catalyst facilitates efficient C–C bond formation between a wide range of π-neutral and π-deficient aryl boronic acids and N-acyliminium precursors derived from quinoline and isoquinoline. The reaction proceeds under mild conditions and is tolerant of common organic functional groups. In addition, a simple one-pot protocol amenable to the direct use of substituted quinolines is described. Consistent with preliminary data revealing a mild, organoboronate-mediated racemization of enantioenriched N-acyliminium precursors, initial results indicate that a chiral phosphoramidite-ligated nickel catalyst promotes enantioselective arylation of racemic substrate with no evidence of a kinetic resolution during catalysis.
Introduction
The intermolecular addition of carbon-centered nucleophiles to iminium and N-acyliminium ions is a powerful strategy for the synthesis of substituted amines that has found widespread use in the rapid assembly of alkaloids and active pharmaceutical ingredients.1 Classically, this transformation has been achieved with strongly nucleophilic organometallic reagents (RMgX, RLi, R2Zn), π-rich arenes (Friedel–Crafts reaction), allylsilanes and stannanes, as well as enolates and their synthetic equivalents (Mannich reaction).2 The seminal work of Petasis expanded the scope of the Mannich reaction by demonstrating that vinyl boronic acids undergo 1,2-addition to iminium ions as part of a three-component coupling.3 Subsequent studies have demonstrated that π-rich aryl and heteroaryl boronic acids also participate in the reaction, but π-neutral and π-deficient aryl boronic acids show little or no reactivity in the absence of directing groups on the substrate.4 Herein, we report a distinct strategy to effect catalytic C–C bond formation with N-acyliminium ions and organoboronates.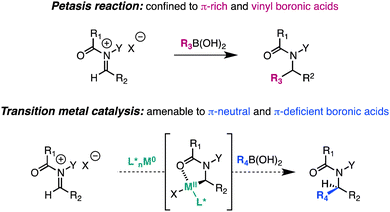
As a first example, we document a Ni(0)-catalyzed Petasis-like transformation between π-neutral and π-deficient aryl boronic acids and quinoline-derived N,O-acetals. Application of this strategy to the three-component coupling of azaaromatics, chloroformates, and aryl boronic acids provides a general route for the construction of 2-arylated dihydroquinolines and 1-arylated dihydroisoquinolines that comprise the core of numerous bioactive agents.5
Pioneering studies by Beller and Arntdsen have documented the ability of coordinatively unsaturated transition metal catalysts to oxidatively insert into N-acyliminium ion precursors.6 Subsequent reports by Arntdsen established that the resulting cyclometalated adducts undergo C–C bond formation with vinyl stannanes as well as carbonylative coupling reactions with vinyl and aryl stannanes.7 Given the intriguing reactivity profile of such complexes, we proposed that N-acyliminium-derived metallacycles might function as intermediates in Suzuki–Miyaura reactions with appropriate organoboronate partners. In contrast to the Petasis reaction, transition metal-catalyzed cross-coupling methodologies have progressed to such a degree that high efficiency is achieved nearly independently of the electronic nature and structure of the organometallic reagent. Consequently, we envisioned that this approach would expand conventional iminium additions to those previously inaccessible by Friedel–Crafts chemistry, allowing stable and widely available aryl, vinyl, and alkyl boronates to function as competent nucleophiles under mild and functional group-tolerant conditions.8 In addition, this approach could provide a common mechanism amenable to the development of catalyst-controlled enantioselective C–C bond-forming reactions, and in so doing, represents a strategic alternative for the asymmetric catalytic synthesis of nitrogen-containing heterocycles.9
Results and discussion
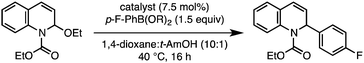
To test this proposal, commercially available 2-ethoxy-1-ethoxycarbonyl-1,2-dihydroquinoline (EEDQ) was selected as a substrate for initial catalyst screening studies.10N,O-Acetals such as EEDQ serve as convenient N-acyliminium ion precursors that can be revealed by mild Lewis or Brønsted acid additives.1 We surmised that the Lewis acidic nature of the boronic acids11 might be sufficient to (1) facilitate oxidative addition12 of a low valent metal to the N,O-acetal substrate and (2) simultaneously activate the boronic acid toward transmetalation via formation of an ate complex.13Transition-metal complexes typically employed in imine arylation14 and Suzuki cross coupling15 were evaluated for their ability to promote C–C bond formation between EEDQ and p-fluorophenyl boronic acid (p-F-PhB(OH)2) at 40 °C (Table 1). Notably, less than 5% product was formed in the absence of a transition metal catalyst after 16 h, unambiguously demonstrating the limitations of the traditional Petasis borono–Mannich reaction (Table 1, entry 1). In keeping with the ability of [Rh]–Ar and [Ir]–Ar intermediates to undergo efficient conjugate addition,16 reactions with [Rh(cod)Cl]2 and [Ir(cod)Cl]2 gave the undesired 1,4-dihydroquinoline regioisomer in >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 rr,17 albeit in low yield (Table 1, entries 2 and 3). In contrast, a Pd(0) catalyst provided the dihydroquinoline product in an improved 52% yield and with high 1,2-regioselectivity, consistent with the intermediacy of a chelated intermediate (Table 1, entry 4). Among the other transition metal catalysts examined, Ni(cod)2 with equimolar PPh3 (7.5 mol% each) proved optimal (Table 1, entry 6).18 With this catalyst system, the desired product was isolated in 94% yield and >20:1 rr when the cyclic anhydride of the boronic acid (aryl boroxine = (ArBO)3) was employed (Table 1, entry 7).19 Since EEDQ is susceptible to decomposition by hydrolysis,20 use of the boroxine leads to higher reaction efficiency at least in part by minimizing the amount of H2O introduced to the reaction.
:1 rr,17 albeit in low yield (Table 1, entries 2 and 3). In contrast, a Pd(0) catalyst provided the dihydroquinoline product in an improved 52% yield and with high 1,2-regioselectivity, consistent with the intermediacy of a chelated intermediate (Table 1, entry 4). Among the other transition metal catalysts examined, Ni(cod)2 with equimolar PPh3 (7.5 mol% each) proved optimal (Table 1, entry 6).18 With this catalyst system, the desired product was isolated in 94% yield and >20:1 rr when the cyclic anhydride of the boronic acid (aryl boroxine = (ArBO)3) was employed (Table 1, entry 7).19 Since EEDQ is susceptible to decomposition by hydrolysis,20 use of the boroxine leads to higher reaction efficiency at least in part by minimizing the amount of H2O introduced to the reaction.
| Entry | Catalyst | Nucleophile | Yield (%) | rrb |
|---|---|---|---|---|
|
a See supporting information for details.† Yields determined by 19F NMR using PhCF3 as a quantitative internal standard.
b 1,2:1,4 regioisomeric ratio; determined by 19F NMR analysis of the crude reaction mixture.
c Reactions run at 60 °C, 80 °C, and 100 °C provide similar results.
d Isolated yield after column chromatography.
e Using 0.5 equiv of (p-F-PhBO)3.
|
||||
| 1 | None | p-F-PhB(OH)2 | <5c | — |
| 2 | [Rh(cod)Cl]2 | p-F-PhB(OH)2 | 24 | 1:>20 |
| 3 | [Ir(cod)Cl]2 | p-F-PhB(OH)2 | 9 | 1:>20 |
| 4 |
Pd2(dba)3/PPh3 (1:1) |
p-F-PhB(OH)2 | 52 | >20:1 |
| 5 | Ni(PPh3)2Cl2 | p-F-PhB(OH)2 | <5 | — |
| 6 |
Ni(cod)2/PPh3 (1:1) |
p-F-PhB(OH)2 | 74d | >20:1 |
| 7 |
Ni(cod)2/PPh3 (1:1) |
(p-F-PhBO)3e | 94d | >20:1 |
These optimized conditions are general for aryl-boroxines containing substitution at the ortho, meta, and para positions (Scheme 1). In the case of π-neutral aryl-boroxines, near-quantitative yields are obtained for the cross coupling. Perhaps most important, the reaction proceeds well with electron-deficient boronic acids that are traditionally inert to Petasis conditions (Scheme 1, 55–95% yield). Hindered boronic acids are also competent reaction partners, although C–C bond formation takes place with reduced efficiency (e.g. o-tolyl boroxine: 63% yield). On the other hand, our initial attempts using π-deficient heteroaryl boroxines (e.g. 3-pyridyl boroxine), challenging partners for Csp2–Csp2 Suzuki cross couplings,21 led to no reaction. In all cases, exclusion of the Ni(0) catalyst provides minimal product (<5%).
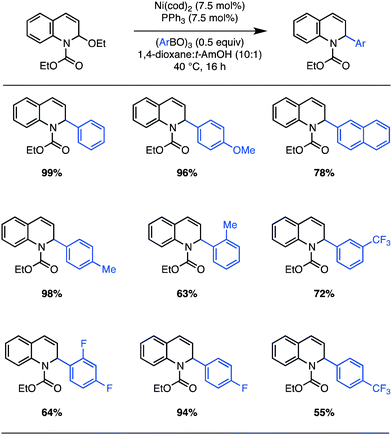 | ||
| Scheme 1 Ni(0)-catalyzed arylation of EEDQ: scope. Isolated yields after column chromatography for reactions run on 0.50 mmol scale (average of two runs). | ||
Considering the value of quinoline-derived intermediates in complex target and bioactive molecule synthesis,22 we sought a simple, one-pot protocol amenable to the direct use of substituted quinolines (Scheme 2). Treatment of quinoline with excess ethyl chloroformate, MeOH, and Na2CO3,23 followed by exposure to the optimized nickel-catalyzed conditions, provided the dihydroquinoline cross-coupling product in 95% yield (Scheme 2; compared with 99% from EEDQ in Scheme 1).24
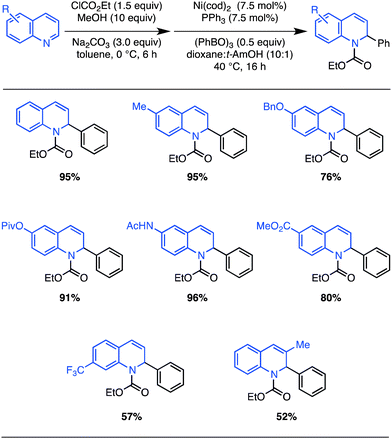 | ||
| Scheme 2 One-pot N,O-acetal formation and Ni(0)-catalyzed arylation of substituted quinolines: scope. Isolated yields after column chromatography for reactions run on 0.50 mmol scale (average of two runs). | ||
A survey of the scope of this in situ protocol was conducted, revealing that quinolines substituted with common functional groups at the 6-position are well tolerated (Scheme 2). However, quinolines substituted at the 7-position with strong electron-withdrawing groups or at the 3-position with alkyl groups react with reduced efficiency. The efficient formation of dihydroquinolines bearing amides and esters is worthy of direct mention since their preparation using strongly nucleophilic organomagnesium or organolithium reagents would likely not be successful. Notably, in all cases, the dihydroquinoline products are obtained in >20:1 regioselectivity whereas mixtures of regioisomers typically result from uncatalyzed reactions with stronger nucleophiles.
We next tested whether the reaction could be extended to the arylation of other azaaromatic substrates. Although it was necessary to increase the catalyst loading (15 mol%) and equivalents of phenyl boroxine (0.75 equiv), the reaction with an isoquinoline-derived N,O-acetal proceeded in a promising 71% yield (Scheme 3). This result provides a preliminary indication of the utility of the catalytic strategy to effect addition reactions with an array of N-acyliminium ion precursors.
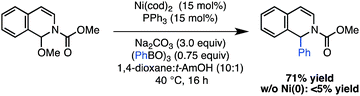 | ||
| Scheme 3 Application of Ni(0)-catalyzed arylation to an isoquinoline derived N,O-acetal. | ||
A preliminary proposal for the mechanism of the nickel-catalyzed coupling is presented in Scheme 4. As a working hypothesis, we suggest that the boroxine coupling partner facilitates addition of Ni(0) to EEDQ through ionization of the N,O-acetal, affording a Ni(II) complex A,25,26 analogous to the palladium complexes characterized by Arndtsen.6d According to a traditional cross-coupling cycle, transmetalation between A and the boronic acid ate complex to give B, followed by reductive elimination, would deliver the arylated product and complete catalyst turnover. An alternative mechanism in which the nickel catalyst undergoes direct oxidative addition to the allylic ether of EEDQ and the benzylic ether of the isoquinolineN,O-acetal (Scheme 3) is also plausible in light of related studies on nickel-catalyzed substitutions with allylic ethers.27 However, a few pieces of information suggest a role for the boroxine as a Lewis acid activator of the C–O bond of the N,O-acetal. The use of phenyl boronate esters and phenyl trifluoroborate28 as nucleophiles in place of phenyl boroxine/boronic acid led to no reaction, consistent with their reduced Lewis acidity. In addition, preliminary NMR experiments have shown that the nickel catalyst does not undergo oxidative addition to EEDQ in the absence of additives.
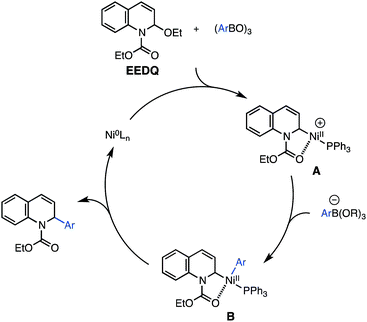 | ||
| Scheme 4 Proposed mechanism. | ||
To further test this proposed mechanism, enantioenriched EEDQ (84% ee) was subjected to the nickel-catalyzed cross-coupling conditions with phenyl boroxine; in accord with the intermediacy of an N-acyliminium ion and/or a configurationally unstable organonickel complex, the dihydroquinoline product was formed as a racemic mixture. Similarly, exposure of EEDQ (84% ee)29 to phenyl boroxine in the absence of the nickel catalyst led to racemization of EEDQ over 5 h. Importantly, these observations suggest that attempts at rendering the coupling reaction enantioselective will not be complicated by kinetic resolution of the racemic starting material. Indeed, we have obtained promising preliminary evidence that a chiral catalyst imparts modest levels of asymmetry within the coupling protocol (Scheme 5). Use of the chiral phosphoramiditeligand, Monophos, in place of PPh3 led to the efficient room temperature coupling of phenyl boroxine and EEDQ in 52% ee.29 Notably, this example constitutes the first reported catalytic asymmetric arylation of N-acylquinolinium ions.9a
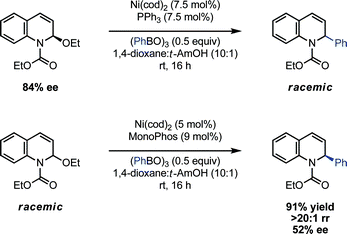 | ||
| Scheme 5 Asymmetric Ni(0)-catalyzed arylation of EEDQ. | ||
Conclusions
In summary, we have developed a Ni(0)-catalyzed reaction that facilitates efficient coupling between a wide range of π-neutral and π-deficient aryl boronic acids and N-acyliminium precursors derived from quinoline and isoquinoline. Preliminary studies indicate a mechanism involving organoboronate-mediated activation of EEDQ and support the viability of rendering the process enantioselective. Application of the protocol to related substrate classes and further development of an enantioselective method are ongoing and will be reported in due course.Acknowledgements
We thank Dr. Pat Carroll of the University of Pennsylvania for X-ray crystallographic structural determination and Lotus Separations for chiral separation of EEDQ. Financial support was provided by Princeton University and kind gifts from Eli Lilly and Sanofi-Aventis. The authors also thank the MacMillan and Sorensen groups for use of their chemicals and instruments.Notes and references
- (a) A. Yazici and S. G. Pyne, Synthesis, 2009, 339 CAS; (b) A. Yazici and S. G. Pyne, Synthesis, 2009, 513 CAS; (c) W. N. Speckamp and M. J. Moolenaar, Tetrahedron, 2000, 56, 3817 CrossRef CAS.
- For a review, see: M. Arend, B. Westermann and N. Risch, Angew. Chem., Int. Ed., 1998, 37, 1044 Search PubMed.
- (a) N. A. Petasis and I. Akritopoulou, Tetrahedron Lett., 1993, 34, 583 CrossRef; (b) N. A. Petasis and I. A. Zavialov, J. Am. Chem. Soc., 1997, 119, 445 CrossRef CAS; (c) N. A. Petasis, A. Goodman and I. A. Zavialov, Tetrahedron, 1997, 53, 16463 CrossRef CAS; (d) N. A. Petasis and I. A. Zavialov, J. Am. Chem. Soc., 1998, 120, 11798 CrossRef CAS.
- For a review, see: (a) N. R. Candeias, F. Montalbano, P. M. S. D. Cal and P. M. P. Gois, Chem. Rev., 2010, 110, 6169 CrossRef CAS; (b) for an example, see: R. A. Batey, D. B. MacKay and V. Santhakumar, J. Am. Chem. Soc., 1999, 121, 5075 Search PubMed.
- For a review, see: (a) M. E. Welsch, S. A. Snyder and B. R. Stockwell, Curr. Opin. Chem. Biol., 2010, 14, 347 CrossRef CAS.
- For a review, see: (a) M. Beller and M. Eckert, Angew. Chem., Int. Ed., 2000, 39, 1010 CrossRef CAS. For examples, see (b) R. D. Dghaym, K. J. Yaccato and B. A. Arndtsen, Organometallics, 1998, 17, 4 CrossRef CAS; (c) J. L. Davis and B. A. Arndtsen, Organometallics, 2000, 19, 4657 CrossRef CAS; (d) Y. Lu and B. A. Arndtsen, Org. Lett., 2007, 9, 4395 CrossRef CAS.
- (a) J. L. Davis, R. Dhawan and B. A. Arndtsen, Angew. Chem., Int. Ed., 2004, 43, 590 CrossRef CAS; (b) A. R. Siamaki, D. A. Black and B. A. Arndtsen, J. Org. Chem., 2008, 73, 1135 CrossRef CAS.
- Arndtsen and co-workers have identified a copper catalyst for mild arylation, vinylation, and alkylation of N-acyliminium ions with organoindium reagents: (a) D. A. Black and B. A. Arndtsen, Org. Lett., 2006, 8, 1991 CrossRef CAS; (b) R. E. Beveridge, D. A. Black and B. A. Arndtsen, Eur. J. Org. Chem., 2010, 3650 CrossRef CAS.
- For a recent review, see: (a) M. Ahamed and M. H. Todd, Eur. J. Org. Chem., 2010, 5935 CrossRef CAS. For relevant examples of catalytic additions to N-acyl quinolinium ions, see: (b) M. Takamura, K. Funabashi, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2000, 122, 6327 CrossRef CAS; (c) Y. Yamaoka, H. Miyabe and Y. Takemoto, J. Am. Chem. Soc., 2007, 129, 6686 CrossRef CAS. For addition to N-acyl isoquinolinium ions, see: (d) M. S. Taylor, N. Tokunaga and E. N. Jacobsen, Angew. Chem., Int. Ed., 2005, 44, 6700 CrossRef CAS. For addition to N-acyl pyridinium ions, see: (e) E. Ichikawa, M. Suzuki, K. Yabu, M. Albert, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2004, 126, 11808 CrossRef CAS; (f) D. A. Black, R. E. Beveridge and B. A. Arndtsen, J. Org. Chem., 2008, 73, 1906 CrossRef CAS; (g) M. A. Fernandez-Ibanez, B. Macia, M. G. Pizzuti, A. J. Minnaard and B. L. Feringa, Angew. Chem., Int. Ed., 2009, 48, 9339 CrossRef CAS.
- Y. M. Chang, S. H. Lee, M. H. Nam, M. Y. Cho, Y. S. Park and C. M. Yoon, Tetrahedron Lett., 2005, 46, 3053 CrossRef CAS.
- (a) D. G. Hall, Boronic acids: preparation and applications in organic synthesis and medicine, Wiley-VCH, Weinheim, 2005. Search PubMedFor specific examples, see: (b) K. Ishihara, H. Kurihara and H. Yamamoto, Synlett, 1997, 597 CrossRef CAS; (c) K. Ishihara, S. Ohara and H. Yamamoto, J. Org. Chem., 1996, 61, 4196 CrossRef CAS.
- For a recent example of Lewis acid assisted oxidative addition, see: (a) Y. Hirata, A. Yada, E. Morita, Y. Nakao, T. Hiyama, M. Ohashi and S. Ogoshi, J. Am. Chem. Soc., 2010, 132, 10070 CrossRef CAS; (b) N. M. Brunkan, D. M. Brestensky and W. D. Jones, J. Am. Chem. Soc., 2004, 126, 3627 CrossRef CAS.
- For a recent investigation on the mechanism of arylboronate transmetalation, see: B. P. Carrow and J. F. Hartwig, J. Am. Chem. Soc., 2011 DOI:10.1021/ja1108326.
- For an example, see: (a) T. Hayashi and M. Ishigedani, J. Am. Chem. Soc., 2000, 122, 976 CrossRef CAS. For a review, see: (b) K. Fagnou and M. Lautens, Chem. Rev., 2003, 103, 169 CrossRef CAS.
- For reviews, see: (a) R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461 CrossRef CAS; (b) G. C. Fu, Acc. Chem. Res., 2008, 41, 1555 CrossRef CAS; (c) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS.
- T. Hayashi and K. Yamasaki, Chem. Rev., 2003, 103, 2829 CrossRef CAS.
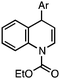
- The 1,4-regioisomer has the structure shown below. See the Supporting Information for characterization data.†
. - Use of other ethereal (DME, Et2O, THF) and alcohol (MeOH, EtOH, i-PrOH, H2O) solvents led to poorer results.
- Aryl boroxines can be readily prepared by heating commercially available boronic acids under reduced pressure. See Supporting Information for details†.
- For a mechanistic study of EEDQ hydrolysis, see: D. J. Cremin, A. F. Hegarty and M. J. Begley, J. Chem. Soc., Perkin Trans. 2, 1980, 412 Search PubMed.
- N. Kudo, M. Perseghini and G. C. Fu, Angew. Chem., Int. Ed., 2006, 45, 1282 CrossRef CAS.
- For reviews of alternative methods to prepare tetrahydro- and dihydroquinolines and isoquinolines, see: (a) M. Chrzanowska and M. D. Rozwadowska, Chem. Rev., 2004, 104, 3341 CrossRef CAS; (b) A. R. Katritzky, S. Rachwal and B. Rachwal, Tetrahedron, 1996, 52, 15031 CrossRef CAS.
- For a related procedure to prepare N,O-acetals, see: P. Bergthaller, German Patent 2502201, 1976.
- The nickel-catalyzed reaction proceeds with comparable efficiency for the synthesis of Cbz- and Fmoc-protected dihydroquinoline products.
- T. Yamamoto, J. Ishizu and A. Yamamoto, J. Am. Chem. Soc., 1981, 103, 6863 CrossRef CAS.
- Under the conditions described in Table 1, Ni(PPh3)2Cl2 does not undergo reduction to Ni(0) (entry 7). The observation that the Ni(II) complex is unreactive suggests that the active catalyst is not serving the role of a Lewis acid nor a nucleophile delivery agent.
- It is important to note that direct oxidative additions to alkyl allylic ethers such as EEDQ, in contrast to reactions with allylic acetates or phenolates, typically require the use of a strong Lewis acid activator, high reaction temperatures or strongly nucleophilic coupling partners such as Grignard reagents: (a) T. Yamamoto, J. Ishizu and A. Yamamoto, Chem. Lett., 1979, 1385 CrossRef CAS; (b) N. Nomura and T. V. RajanBabu, Tetrahedron Lett., 1997, 38, 1713 CrossRef CAS; (c) M. T. Didiuk, J. P. Morken and A. H. Hoveyda, J. Am. Chem. Soc., 1995, 117, 7273 CrossRef CAS; (d) H. Tsukamoto, T. Uchiyarna, T. Suzuki and Y. Kondo, Org. Biomol. Chem., 2008, 6, 3005 RSC; (e) R. Matsubara and T. F. Jamison, J. Am. Chem. Soc., 2010, 132, 6880 CrossRef CAS; (f) T. Nishikata and B. H. Lipshutz, J. Am. Chem. Soc., 2009, 131, 12103 CrossRef CAS; (g) Y. Lu and B. A. Arndtsen, Angew. Chem., Int. Ed., 2008, 47, 5430 CrossRef CAS.
- Potassium phenyltrifluoroborate, phenylboronic acid 1,3-propanediol ester, and phenylboronic acidpinacolester failed to give the desired product.
- The absolute stereochemistry has not been confirmed at this time.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, details of optimization studies, and spectroscopic data for all new compounds. CCDC reference number 808622. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00026h |
| This journal is © The Royal Society of Chemistry 2011 |