A hybrid magnet with coexistence of ferromagnetism and photoinduced Fe(III) spin-crossover†
Miguel
Clemente-León
*a,
Eugenio
Coronado
*a,
Maurici
López-Jordà
a,
Cédric
Desplanches
b,
Saket
Asthana
b,
Hongfeng
Wang
b and
Jean-François
Létard
b
aInstituto de Ciencia Molecular, Universidad de Valencia, Catedrático José Beltrán 2, Paterna, 46980, Spain. E-mail: miguel.clemente@uv.es; Fax: +34 963543273; Tel: +34 963544405eugenio.coronado@uv.es
bCNRS, Université de Bordeaux, ICMCB, 87 avenue du Dr A. Schweitzer, Pessac, F-33608, France
First published on 5th April 2011
Abstract
The insertion of a [Fe(sal2-trien)]+ complex cation into a 2D oxalate network results in a hybrid magnet with coexistence of magnetic ordering and photoinduced spin-crossover (LIESST effect) in compound [FeIII(sal2-trien)][MnIICrIII(ox)3]·(CH2Cl2) (1). A complete photomagnetic characterization together with a detailed structural analysis of the low-spin (LS) and high-spin (HS) structures of 1 is presented in order to understand such unusual behavior. This very rare and unexpected property in a FeIII spin-crossover complex, has been attributed to the strong distortion exhibited by the metastable HS state. Furthermore, 1 has shown that, in contrast to what has been previously proposed, a cooperative spin-crossover is not a necessary condition to observe LIESST effect in FeIII compounds. The photo-induced spin conversion of the inserted FeIII complex has shown to have a negligible influence on the cooperative magnetic behaviour of the 2D oxalate network.
Introduction
Multifunctionality is one of the most appealing topics in chemical science. A rational approach to design multifunctional materials consists of building up two-network hybrid solids formed by two molecular fragments where each network furnishes distinct physical properties. If the two networks are quasi-independent, a coexistence of the two physical properties is anticipated.1 If the two molecular networks are coupled, an interplay between their properties may be observed.1b Finally, if one of these two networks is physically (or chemically) responsive (i.e., if it changes its properties under the application of an external stimulus, like for example light, pressure or temperature), a switchable multifunctional material can be obtained in which the responsive network can influence the properties of the other network.2Bimetallic oxalate-bridged complexes of formula A[MIIMIII(ox)3] (MIII = Cr, Fe, Ru, V, Mn; MII = Mn, Fe, Co, Ni, Cu, Zn; A = Cation; ox = oxalate) have provided remarkable examples of these types of materials.3 They are composed by polymeric 2D4 or 3D5 anionic networks, which furnish the cooperative magnetic properties (ferro-, ferri- or canted antiferromagnetism), and a bulky charge-compensating molecular cation, which templates the network formation and adds a second physical property to the solid. The insertion of different cations into oxalate networks has led to compounds combining the long-range magnetic ordering from the oxalate network with paramagnetism,6 photochromism,7 electric conductivity,8proton conduction,9 chirality10 or very recently, chirality and conductivity in the same compound.11
The interplay between the properties of the two molecular networks has been observed, for example, in enantiopure 2D oxalate-based magnets prepared by Train et al. that exhibit magneto-chiral dichroic effect and magnetisation-induced second harmonic generation.12
The design of switchable magnets has been less explored. A promising approach to reach this goal consists of combining the bimetallic oxalate network with spin-crossover complexes. These kinds of molecular complexes change their spin state from low-spin (LS) to high-spin (HS) configurations when submitted to an external stimulus such as temperature, light-irradiation or pressure. As this magnetic switching is accompanied by changes in the molecular size, the spin-crossover process should act as an internal pressure in the hybrid material and therefore, it might affect the long-range magnetic ordering in the extended magnetic network.
Although several compounds combining spin-crossover cations and bimetallic oxalate anion networks have been reported,13,14 the only one showing coexistence of spin-crossover and ferromagnetism is [FeIII(sal2-trien)][MnIICrIII(ox)3]·(CH2Cl2) (1) reported very recently by our group.14 This compound is formed by [Fe(sal2-trien)]+ cations inserted between MnIICrIII bimetallic oxalate layers. It presents a gradual spin-crossover transition from 350 to 160 K, and a ferromagnetic ordering at 5.6 K. Furthermore, by changing the synthetic conditions a second compound of formula [FeIII(sal2-trien)][MnIICrIII(ox)3]·(CH3OH) (2) has also been obtained. This compound shows an unusual non-chiral 3D network with inserted [Fe(sal2-trien)]+ cations, exhibiting a gradual spin-crossover of 30% of the Fe(III) from 300 to 130 K, together with a ferromagnetic ordering at 5.2 K. Unfortunately, in these compounds no interplay between these two properties—spin-crossover and ferromagnetism—has been detected. This is probably due to the large difference in temperature at which the two phenomena occur. Thus, while the spin-crossover occurs at high temperatures (in the range 130–350 K), the ferromagnetic ordering occurs below 5.2–5.6 K.
A way to overcome this limitation is to take advantage of the photo-induced spin-crossover effect, also known as LIESST effect (light-induced excited spin-state trapping).15 This phenomenon was originally established for FeII complexes at low-temperatures (typically below 50 K).16 In principle, the relaxation via a tunneling process from the HS to the LS state is much faster in FeIII compounds because the changes in the metal–ligand bond lengths between the two states are significantly smaller. However, there are two examples of FeIII complexes showing the LIESST effect reported by Sato et al.17,18 The explanation given by these authors for such unexpected behavior is related to the large structural distortion between the LS and HS states, with the intermolecular interactions playing an important role in the relaxation behavior.18
In this work, we will explore the possibility of using light irradiation to induce spin-crossover in these hybrid materials. We will show that the confinement of [Fe(sal2-trien)]+ cations provided by the 2D bimetallic oxalate layers in 1 can also induce LIESST effect in FeIII complexes. A complete photomagnetic characterization together with a detailed structural analysis of the LS and HS structures of 1 is presented in order to understand such unusual behavior. Finally, the possibility of using this family of compounds as switching magnets will be explored.
Results
Crystal structure of 1
The syntheses, crystal structures, Mössbauer spectroscopy and magnetic properties of 1 and 2 were reported previously.14 In this work, four crystal structures of 1 at 92, 120, 250 and 300 K were solved to study the structural changes between the LS and HS structures.19 Furthermore, we used for comparison the structural data at 180 K from the previous work14 and measured the unit cell of one crystal between 92 and 290 K.The structure of 1 is formed by anionic [MnCr(ox)3]− sheets in the bc plane alternating with inter-lamellar [Fe(sal2-trien)]+ cations and dichloromethane solvent molecules (see Fig. 1 and Fig. 2).
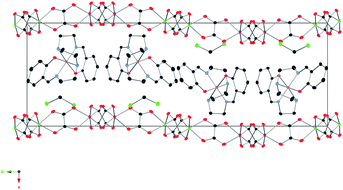 | ||
| Fig. 1 Projection of the structure of 1 in the ab plane at 120 K. Fe (yellow), Cl (green) C (black), N (blue), O (red) Mn(pink), Cr(green). Hydrogen atoms have been omitted for clarity. | ||
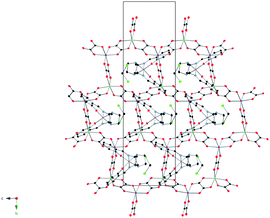 | ||
| Fig. 2 Projection of the structure of 1 in the bc plane at 120 K. Fe (yellow), Cl (green) C (black), N (blue), O (red) Mn(pink), Cr(green). Hydrogen atoms have been omitted for clarity. | ||
To evaluate the structural distortion between the LS and HS octahedral coordination geometries of [Fe(sal2-trien)]+ in 1, we have focused on the differences observed in the crystal structures measured at 120 and 300 K for the same crystal because they correspond roughly to the LS and HS structures.
The first difference is a shortening of metal–ligand bond lengths as expected for a spin-crossover. Thus, the average Fe–N and Fe–O bond distances in the structure solved at 300 K are respectively, 2.124(4) and 1.890(4) Å, while those solved at 120 K are 1.975(2) and 1.866(2) Å. This results in average bond length differences ΔrHL,Fe–N = 0.149 Å and ΔrHL,Fe–O = 0.024 Å.
The second difference concerns the distortion of the octahedral coordination site, which is significantly larger for the HS state. Thus, at 300 K two of the three diagonals from the octahedron, defined by the bonds N(amine)–Fe–O(phenoxo) (N(amine) = N2,N3), differ strongly from 180° (158.08(19) and 161.23(15)°), while the remaining one, defined by the bond N(imine)–Fe–N(imine), is 176.78(15)° (N(imine) = N1,N4). On the contrary, the coordination octahedron is much more regular at 120 K. The three diagonals are much closer to 180° with N(amine)–Fe–O(phenoxo) angles of 173.18(10) and 173.82(9)° and N(imine)–Fe–N(imine) angle of 179.63(11)°. Different parameters have been used by several authors to quantify the distortions of [Fe(sal2-trien)]+ salts, such as Σ and Θ (see Table 1).20–22 As expected, angular and trigonal distortions, which are evaluated respectively by Σ and Θ, are much more important when the FeIII complex is in the HS state (300 K). On the other hand, Σ and Θ parameters of the LS structures (92, 120 and 180 K) present very similar values while the structure with intermediate spin state (250 K) present intermediate values between the LS and HS structures as expected (see Table 1).
| Spin state | Mean Fe–N (Å) | Mean Fe–O (Å) | Σ (°)a | Θ (°)b | |
|---|---|---|---|---|---|
a
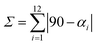 where αi are the 12 cis-N/O–Fe–N/O angles around the iron atom.
b where αi are the 12 cis-N/O–Fe–N/O angles around the iron atom.
b
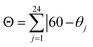 where θj are the 24 unique N/O–Fe–N/O angles measured on the projection of two triangular faces of the octahedron along their common pseudo-threefold axis. where θj are the 24 unique N/O–Fe–N/O angles measured on the projection of two triangular faces of the octahedron along their common pseudo-threefold axis.
|
|||||
| 1 (300 K) | ∼ 80% HS | 2.124(4) | 1.890(4) | 88 | 244 |
| 1 (250 K) | ∼ 50% HS | 2.076(4) | 1.871(3) | 72 | 217 |
| 1 (180 K) | ∼ 90% LS | 1.989(4) | 1.866(3) | 52 | 89 |
| 1 (120 K) | LS | 1.975(2) | 1.866(2) | 49 | 87 |
| 1 (92 K) | LS | 1.978(3) | 1.864(2) | 50 | 86 |
| 2 (120 K) [Fe1] | ∼ 72% HS | 2.039(15) | 1.886(12) | 71 | 190 |
| [Fe2] | 2.14(2) | 1.859(14) | 89 | 235 |
The third difference concerns the structural disorder shown by one of the ethylene groups at 300 K, which can be solved by considering two possible configurations with occupancies of 58 and 42%. This disorder is not observed in the structures close to the LS state at 180, 120 and 92 K but it is maintained in the structure with ∼50% of FeIII in the HS state (250 K) with occupancies of 56 and 44%. Dichloromethane molecules that occupy the holes left by the [Fe(sal2-trien)]+ cations and the oxalate network, present a similar disorder at 250 and 300 K, which disappears at 92, 120 and 180 K.
The crystal packing of [Fe(sal2-trien)]+ complexes between the low and high temperature structures also presents some changes, which are reflected in the lattice parameters. Changes in the unit cell of 1 indicate a shortening of a and b axis from 300 to 92 K whereas the c axis remains almost constant with a small increase at decreasing temperature (see Fig. S1†). Furthermore, the β angle decreases gradually from 300 to 92 K. All these changes are more important in the spin-crossover region from 300 to 175 K indicating that they are associated with the change of spin state of [Fe(sal2-trien)]+. To understand these changes, let us first describe the intermolecular contacts between the FeIII complexes. In the present case, this packing is restricted in the bc plane as the FeIII complexes are confined between two anionic bimetallic oxalate layers. They lie with their longer axis parallel to the oxalate layers, and they are connected through edge-to-face C–H⋯π and N–H⋯π interactions23 (see Figs. S2 and S3†) forming double chains running along the c axis. From the two phenoxy rings of sal2-trien, only the one placed in the internal part of the double chain is involved in these C–H⋯π and N–H⋯π interactions with four adjacent FeIII complexes belonging to the same double chain. Furthermore, the phenoxy ring belonging to different double chains present π–π stacking interactions. Finally, there are numerous short contacts with the oxalate ligands of the two neighboring layers (see Figs S4 and S5†).
As the a parameter gives the separation between the oxalate layers (from 12.096(5) Å at 300 K to 11.5561(3) Å at 120 K), the shortening of a axis arises from the change in the spin state of the FeIII complex. The most important consequence of this shortening is that at 120 K there are shorter contacts between O atoms from oxalate layers and CH groups from [Fe (sal2-trien)]+ (see SI†). On the other hand, the most important effect of the shortening of b axis (from 32.135(5) Å at 300 K to 31.6369(7) Å at 120 K) is the presence of shorter contacts at 120 K between FeIII complexes belonging to the same double chains through N–H⋯π edge-to-face interactions and between FeIII complexes belonging to different double chains through π–π stacking interactions. On the contrary, offset π–π stacking interactions appear in the structure at 300 K between C atoms of phenoxy rings belonging to different double chains (in green in Figure S2†) with weaker contacts in the structure solved at 120 K (see SI†).
Change in the magnetic properties of 1 upon irradiation
In Fig. 3, we plot the magnetic behavior of 1 before and after light irradiation. Before irradiation χT shows a constant value of 10.0 emu·K mol−1 in the high temperature range (from 400 to 350 K), which is approximately equal to the sum of the expected contributions for the isolated paramagnetic ions with ∼80% of FeIII spin-crossover complex in a HS state. From 350 to 165 K, χT decreases gradually to reach a minimum χT value at 165 K of 7.2 emu·K mol−1. This value is consistent with the Mössbauer data that indicates that FeIII is 94% LS at this temperature.14 Therefore, in this range of temperatures a HS → LS spin conversion of the FeIII complex takes place. Below 165 K, χT increases due to the MnII–CrIII ferromagnetic interactions within the bimetallic oxalate layers. This increase is very sharp at lower temperatures suggesting the onset of long-range ferromagnetic ordering at 5.6 K, as confirmed by AC susceptibility and field cooled (FC) and zero-field cooled (ZFC) magnetization measurements (Figs. S6 and S7†).14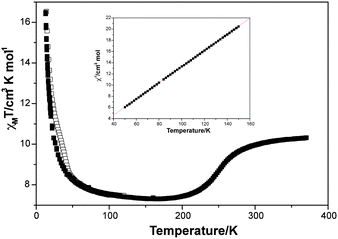 | ||
| Fig. 3 Magnetic and Photo-Magnetic behaviour for compound 1. ■ Magnetic behaviour measured in dark. □ Photo-magnetic behaviour measured after irradiation by 647 nm wavelength laser. Inserted graph: Curie–Weiss law between 50 K and 150 K. | ||
During irradiation at 647 nm wavelength (power 8 mW cm−2), at 10 K inside a SQUID cavity, an increase in χT was observed at low temperatures. Thus, after about 1.5 h the light irradiation was switched off. The temperature was then increased at 0.3 K min−1 and the magnetic susceptibility recorded. Below 50 K, the χT value after irradiation is clearly higher than the one before irradiation, while above 50 K, the two curves are superimposed (Fig. 3). This result suggests that a photomagnetic effect has occurred, which can be attributed to the spin photoconversion of the [Fe(sal2-trien)]+ from its LS ground state at low temperature to its HS metastable state. In order to estimate the fraction of photoconverted FeIII, the magnetism of the MnIICrIII bimetallic layers needs to be substrated as it presents a strong underlying magnetism. To estimate the magnetism of the bimetallic layers we have taken advantage of the fact that below the thermal spin transition (i.e. below 150 K) and well above the Curie temperature (i.e. above 50 K), the magnetic response can be satisfactorily simulated by a Curie–Weiss law (see inset in Fig. 3). Below 50 K, this simple Curie–Weiss law is unable to reproduce properly the χT values due to the vicinity of the ferromagnetic ordering. In this region, we have used the molecular field model proposed by Néel that explicitly takes into account the fact that there are two distinct sub-networks in the MnIICrIII layers.24 Differences between experimental χT values and those obtained with this model are lower than 0.04 cm3 K mol−1 in between 25 K and 175 K. Details of this fitting are given as supporting information.† In Fig. 4, we plot the temperature dependence of the fraction of HS FeIII (γHS) before and after irradiation after subtracting the contribution from the MnIICrIII oxalate network.
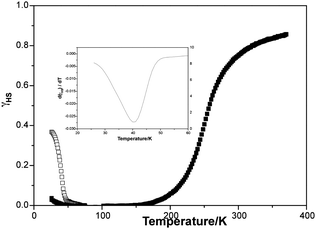 | ||
| Fig. 4 Fraction of HS FeIII (γHS) before (■) and after (□) irradiation). Inserted graph: χT derivative for photo-magnetism. | ||
γ HS is almost zero below 170 K in the measurements performed in the dark. Increasing the temperature above 170 K leads to a gradual increase of γHS with a maximum value of 0.86 at 370 K in agreement with Mössbauer measurements.14γHS from the curve obtained after irradiation reaches a maximum value of 0.37 at 25 K. When the temperature increases, a decrease in γHS is observed, and γHS reaches 0 at about 75 K. At higher temperatures γHS curve is superimposed with that recorded in the dark. The T(LIESST), estimated from the minimum of the derivative of γHS by the temperature (inset in Fig. 4), is determined to be 41 K.25 This represents somewhat the limit of the long-lived photo-induced lifetime of the HS state. In order to get some insight into the decay of the photoinduced HS state, some kinetics have been recorded at 10 K and every 2 K between 36 and 44 K (Fig. 5).
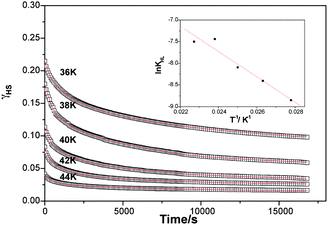 | ||
| Fig. 5 Relaxation kinetics and simulation of the HS fraction vs. time at different temperatures. Inserted graph: lnkHL(T) vs. 1/T dependence. kHLcorresponds to the rate constant for the HS → LS relaxation. The fitting procedure has been performed by adding a small residual HS at long time. | ||
At 10 K, almost no decay of the photo-induced HS state was noticed. More precisely, the decrease of the HS fraction was less than 2% after 7 h. Fig. 5 shows the relaxation kinetics recorded between 36 and 44 K. The maximum of HS fraction at initial time, for a given kinetic is different: the higher the temperature, the lower the photoexcitation. This suggests that the kinetic process is thermally activated. Interestingly, these relaxation curves show strong deviation from a single exponential, with a marked stretched exponential behaviour, and with a fast component at earlier times and a long decay process at infinite time. For reproducing this type of curves, Hauser et al.26 used a distribution of relaxation rates at a given temperature with a Gaussian distribution of the activation energy. Following this procedure, the relaxation curves of compound 1 can be satisfactorily fitted as illustrated by the solid lines in Fig. 5. The apparent activation energy, Ea (= 200 cm−1), and the apparent preexponential factor, k∞ (= 0.49 s−1) of the activated region are calculated from the straight line of the lnkHL(T) vs. 1/T plot. The standard deviation of the activation energy is 52 ± 6 cm−1.
Effect of photoexcitation on the magnetic ordering
FC and ZFC magnetization (Fig. S6†) and AC susceptibility measurements (Fig. S7†) have been recorded between 2 and 10 K, before and after photoirradiation of 1.5 h at 10 K to test if the photoconversion of the FeIII complex affects the Curie temperature (Tc) of the compound. We have to take into account that at 10 K the lifetime of the photoinduced HS state is very long, and consequently the HS fraction remains almost constant during the experiment. ZFC and FC magnetization measurements start to bifurcate at 5.6 K, both before and after irradiation. Similarly, AC susceptibility measurements show the appearance of an out-of-phase signal (χ′′) below Tc = 5.6 K that remains almost constant upon irradiation, showing a shift to higher values by 0.1 K after photoirradiation.Crystal structure and photomagnetism of 2
The structure of 2 was solved at 120 K.14 It is formed by an anionic 3D polymeric oxalate-bridged bimetallic network with [Fe(sal2-trien)]+ cations and methanol solvent molecules occupying the cavities. Magnetic measurements and Mössbauer spectroscopy14 indicate that 100% of FeIII complexes are in the HS at room temperature and the HS → LS spin conversion of 30% of the FeIII complexes from 300 and 130 K. There are two crystallographically independent [Fe(sal2-trien)]+ complexes ([Fe1] and [Fe2]). The average Fe–N and Fe–O bond distances are close to those obtained for other high-spin (HS) [Fe(sal2-trien)]+ compounds (see Table 1). At lower temperatures, the HS fraction remains constant and there is a ferromagnetic ordering with a Tc of 5.2 K, as usual for this type of compounds. In contrast to 1, this compound does not present LIESST effect.Discussion
The lifetime of the HS photoexcited state has been rationalized by Hauser,27 on the basis of a non-adiabatic multiphonon relaxation model. In the framework of this theory, the metal–ligand distance is taken as the reaction coordinate, related to only one vibrational mode, namely the completely symmetrical one. This is the so-called SCC (single configurational coordinate) approach. Based on this theory, it is possible to estimate theoretically the rate constant for the HS → LS relaxation (kHL) at different temperatures for the different ions that could exhibit the LIESST effect such as FeII, FeIII or CoII.28 The main difference between these three ions is the bond length difference between the LS and the HS states, being typically 0.2 Å for FeII, 0.13–0.16 Å for FeIII and 0.09–0.12 Å for CoII. This change in bond length difference is expected to directly affect the decay of the photo-induced state, being much faster for CoII than for FeII and FeIII. Particularly, according to SCC theory, the low-temperature (tunneling regime) lifetime of the HS state for FeIII spin-crossover compounds is expected to be of the order of milliseconds, whereas it can go from seconds to months for FeII spin-crossover compounds. According to such a short lifetime, it should be impossible to observe the decay of the photoinduced state for FeIII by using standard SQUID techniques. However, as mentioned above, the LIESST effect has been observed by Sato in two FeIII complexes, [Fe(pap)2]+ and [Fe(qsal)2]+.18 This author attributed such an extraordinary result to the strong distortion of the FeIII site from its octahedral geometry, which can be enhanced by the presence of strong intermolecular interactions, such as π–π stacking. Thus, when passing from the ideal octahedral site [FeIIN6] to a distorted [FeIIIN4O2] geometry, in addition to the completely symmetrical vibrational mode, other vibrational modes such as the bending mode should also be taken into account.18,29 This results in a displacement between the potential walls of the HS and LS states, which is larger than that expected considering only the changes in metal–ligand distances, and could explain the long lifetime of the photogenerated HS state.To evaluate the distortion of [Fe(sal2-trien)]+ in 1, we have focused our attention on the differences between the 120 and 300 K structures of the compound as they are close to the LS and HS structures. The structure at 120 K corresponds to the LS structure since magnetic and Mössbauer data indicate that 100% of the FeIII are in the LS state at this temperature. On the other hand, the structure at 300 K is close to the HS structure as magnetic data indicate that around 80% of FeIII are in the HS state at this temperature.14
The first difference between the two structures is a shortening in the Fe–N and Fe–O bond lengths as expected for a spin-crossover. Thus, the average bond length differences are ΔrHL,Fe–N = 0.149 Å and ΔrHL,Fe–O = 0.024 Å. If we take into account that at 300 K about 20% of FeIII remains in the LS state, we could expect ΔrHL,Fe–N = 0.186 Å and ΔrHL,Fe–O = 0.03 Å values between the LS and HS states of this compound. These values are very close to those exhibited by the two FeIII compounds known to date presenting LIESST effect (in [Fe(pap)2]X, ΔrHL,Fe–N = 0.19 Å and ΔrHL,Fe–O = 0.05 Å, and in [Fe(qsal)2]Y, ΔrHL,Fe–N = 0.18 Å and ΔrHL,Fe–O = 0.02 Å).18 Notice that these values are also similar to those reported for [Fe(sal2-trien)][Ni(dmit)2] (ΔrHL,Fe–N = 0.188 Å and ΔrHL,Fe–O = 0.036 Å)30 which is the only other [Fe(sal2-trien)]+ salt reported so far for which both the LS and HS structural data are available.31 This salt is the first example of [Fe(sal2-trien)]+ salt presenting a strong cooperative transition and wide hysteresis but no LIESST effect has been reported for this compound. Furthermore, the synthesis of this compound presents serious problems of reproducibility that could limit further studies.32
A second set of parameters accounting for angular and trigonal distortions from the ideal octahedron is provided by Σ and Θ parameters. However, Σ and Θ values of the 300 and 120 K structures lie in the range of those found for other HS and LS [Fe(sal2-trien)]+ compounds.31 Therefore, it is difficult to get some conclusions from these parameters.
The third difference concerns the changes in selected bond angles (cis and trans N/O–Fe–N/O bond angles) between the LS and HS structures (Δ∢HL). In 1, a value of Δ∢HL = 93.64° is obtained. This value is intermediate between that of [Fe(pap)2]+ (Δ∢HL = 136.06°), for which LIESST effect is observed, and that of [Fe(acpa)2]+ ((Δ∢HL = 74.32°), which does not present LIESST effect, but a larger value could be expected taking into account that there are still around 20% of FeIII in the LS state in the structure solved at 300 K. Therefore, we can conclude that, in analogy with [Fe(pap)2]+, the strong distortion from the ideal octahedral geometry in 1 is at the origin of the LIESST effect.
In turn, a sharp structural difference between 1 and [Fe(pap)2]+ concerns the nature and dimensionality of the intermolecular interactions. Thus, while in [Fe(pap)2]+ the π–π 2D stacking between pyridine and phenyl rings seems to enhance the structural distortion of the LIESST complex, in 1, π–π interactions between pairs of [Fe(sal2-trien)]+ complexes belonging to different chains coexist with edge-to-face CH–π and NH–π interactions. As a consequence, 1 shows a gradual spin-crossover with no hysteresis, while [Fe(pap)2]+ presents a cooperative spin-crossover with hysteresis. Furthermore, the decay of the photo-induced HS state of the two compounds follows a different relaxation kinetics. Whereas the time dependence of the HS fraction after photoexcitation of [Fe(pap)2]+ can be fitted to a sigmoidal-type curve as a result of self-acceleration of the HS → LS relaxation due to cooperative effects, the one from 1 can be fitted to a multiexponential curve with a fast component at earlier times and a long decay process at infinite times. The broad distribution of activation energies is the cause of this type of relaxation kinetics. These curves are typical of non-cooperative systems that rarely present a single-exponential behavior. It appears in poorly crystalline solids and in some cases it has been related to the presence of disorder in the structure in compounds presenting a cooperative spin-crossover.33 Hence, it could have some relation with the disorder observed in the structure of 1 even it is only observed at high temperature. In any case, we can conclude that both the magnetic behavior and the decay of the photo-induced HS state in 1 indicate that in contrast to the previous examples, cooperativity it is not a necessary condition to have LIESST effect in FeIII compounds.
A second structural difference of 1 with respect to the other FeIII compounds exhibiting LIESST effect is the presence of an anionic extended network (the bimetallic oxalate network) instead of a discrete small counterion. This rigid network could play a similar role in the stabilisation of the metastable HS state of 1 to that played by the strong π–π intermolecular interactions in [Fe(pap)2]+. In favour of this hypothesis are the numerous short contacts between the [Fe(sal2-trien)]+ cations with the oxalate ligands, which are different in the low and high-temperature structures (see SI†). Maybe the interactions with the oxalate network enhance the distortion of the LIESST complex but, at the same time, they prevent a cooperative spin-crossover since they restrict the structural dimensionality to 2. Photomagnetic measurements on salts of [Fe(sal2-trien)]+ and derivatives in different anionic environments are needed to clarify this point. This work is in progress in our group.13d
The question remains why no LIESST has been observed on the remaining 30% HS fraction at 10 K for compound 2. Further work is necessary to answer to such question. For now, it remains unclear if the absence of LIESST is linked to: (a) a distortion effect and/or (b) the difference of rigidity between 2D and 3D networks and/or (c) the small amount of FeIII that can be converted. As far as the distortion (a) is concerned, it would be helpful to compare the geometries of the [Fe(sal2-trien)]+ complexes in the HS and the LS state. Unfortunately, this is not possible as the HS fraction remains at 70% at temperatures as low as 10 K. The influence of the rigidity of the network (b) can be seen in the fact that the contraction of the interlayer distance that follows the spin-crossover in 1 cannot be present in 2 that presents a more isotropic 3D network. Finally, the low amount of low spin fraction at low temperature (c) makes it difficult to observe the possible small increase of magnetic response under light, as it will be difficult to distinguish this increase from the important magnetic signal of the oxalate network. New compounds are to be synthesized, 2D and 3D, in order to have more data to draw conclusions.
Conclusions
In this work we have shown that the insertion of a [Fe(sal2-trien)]+ complex cation into a 2D oxalate network results in a hybrid magnet with coexistence of magnetic ordering and photoinduced spin-crossover in compound 1. The use of a molecular approach has led to the coexistence of two properties—ferromagnetism and photo-induced spin crossover—that normally do not appear together in the same compound. This could open the way to prepare switching magnets. Still, in the present case the photo-induced spin conversion of the inserted FeIII complex has shown to have a negligible influence on the cooperative magnetic behaviour of the 2D oxalate network. This is not an unexpected result, as 2D oxalate networks with very different interlayer separations present similar Tc values. In this sense, hybrid compounds formed by insertion of these spin-crossover cations into a 3D oxalate network are expected to be more suitable, as this magnetic network has shown to be much more sensitive to the influence of an internal pressure. Unfortunately, the compound 2 obtained by insertion of a [Fe(sal2-trien)]+ cation into a 3D oxalate network does not present LIESST effect.Another remarkable result has been the observation of LIESST effect in 1. This very rare and unexpected property in a FeIII spin-crossover complex, has been attributed to the strong distortion exhibited by the metastable HS state. However, 1 has shown that, in contrast to what has been previously proposed, a cooperative spin-crossover is not a necessary condition to observe LIESST effect in FeIII compounds. The presence of an extended anion network able to establish strong intermolecular interactions with the spin-crossover cation could also enhance this distortion. If this was true, the confinement of spin-crossover cations into extended networks could be a suitable strategy to induce LIESST effect on other FeIII cations, or to improve LIESST properties in FeII complexes.
Finally, the discovery that [Fe(sal2-trien)]+ complexes can present LIESST effect opens the way for the use of this compound in the preparation of new multifunctional switchable compounds with other properties different from the magnetic ones. An interesting possibility could be the switching of the conductivity in [Fe(sal2-trien)]+ salts of [Ni(dmit)2] anions already reported in the literature.34
Acknowledgements
Financial support from the EU (SPINMOL ERC Advanced Grant to EC), the Spanish Ministerio de Ciencia e Innovación (Project Consolider-Ingenio in Molecular Nanoscience and projects MAT2007-61584 and CTQ-2008-06720) and the Generalitat Valenciana (Prometeo Program) are gratefully acknowledged.Notes and references
- (a) E. Coronado and P. Day, Chem. Rev., 2004, 104, 5419 CrossRef; (b) E. Coronado, C. Martí-Gastaldo, E. Navarro-Moratalla, A. Ribera, S. J. Blundell and P. J. Baker, Nat. Chem., 2010, 2, 1031 CrossRef CAS.
- (a) E. Coronado, M. C. Giménez-López, G. Levchenko, F. M. Romero, V. García-Baonza, A. Milner and M. Paz-Pasternak, J. Am. Chem. Soc., 2005, 127, 4580 CrossRef CAS; (b) E. Coronado, M. C. Giménez-López, T. Korzeniak, G. Levchenko, F. M. Romero, A. Segura, V. García-Baonza, J. C. Cezar, F. M. F. De Groot, A. Milner and M. Paz-Pasternak, J. Am. Chem. Soc., 2008, 130, 15519 CrossRef CAS.
- M. Clemente-León, E. Coronado, C. Martí-Gastaldo and F. M. Romero, Chem. Soc. Rev., 2011, 40, 473 RSC.
- (a) H. Tamaki, Z. J. Zhong, N. Matsumoto, S. Kida, M. Koikawa, N. Achiwa, Y. Hashimoto and H. Okawa, J. Am. Chem. Soc., 1992, 114, 6974 CrossRef; (b) H. Tamaki, M. Mitsumi, N. Nakamura, N. Matsumoto, S. Kida, H. Okawa and S. Ijima, Chem. Lett., 1992, 1975 CAS; (c) C. Mathonière, S. G. Carling, D. Yuscheng and P. Day, J. Chem. Soc., Chem. Commun., 1994, 1551 RSC; (d) C. Mathonière, J. Nutall, S. G. Carling and P. Day, Inorg. Chem., 1996, 35, 1201 CrossRef CAS; (e) R. Pellaux, H. W. Schmalle, R. Huber, P. Fisher, T. Hauss, B. Ouladdiaf and S. Decurtins, Inorg. Chem., 1997, 36, 2301 CrossRef CAS; (f) E. Coronado, J. R. Galán-Mascarós, C. J. Gómez-García, J. M. Martínez-Agudo, E. Martínez-Ferrero, J. C. Waerenborgh and M. Almeida, J. Solid State Chem., 2001, 159, 391 CrossRef CAS; (g) K. S. Min, A. L. Rhinegold and J. S. Miller, Inorg. Chem., 2005, 44, 8433 CrossRef CAS; (h) E. Coronado, J. R. Galán-Mascarós and C. Martí-Gastaldo, J. Mater. Chem., 2006, 16, 2685 RSC.
- (a) S. Decurtins, H. W. Schmalle, P. Schneuwly and H. R. Oswald, Inorg. Chem., 1993, 32, 1888 CrossRef CAS; (b) S. Decurtins, H. W. Schmalle, P. Schneuwly, J. Ensling and P. Gütlich, J. Am. Chem. Soc., 1994, 116, 9521 CrossRef CAS; (c) M. Hernández-Molina, F. Lloret, C. Ruiz-Pérez and M. Julve, Inorg. Chem., 1998, 37, 4141; (d) E. Coronado, J. R. Galán-Mascarós, C. J. Gómez-García and J. M. Martínez-Agudo, Inorg. Chem., 2001, 40, 113 CrossRef CAS; (e) F. Pointillart, C. Train, M. Gruselle, F. Villain, H. W. Schmalle, D. Talbot, P. Gredin, S. Decurtins and M. Verdaguer, Chem. Mater., 2004, 16, 832 CrossRef CAS; (f) M. Clemente-León, E. Coronado, C. J. Gómez-García and A. Soriano-Portillo, Inorg. Chem., 2006, 45, 5653 CrossRef CAS.
- (a) M. Clemente-León, J. R. Galán-Mascarós and C. J. Gómez-García, Chem. Commun., 1997, 1727 RSC; (b) E. Coronado, J. R. Galán-Mascarós, C. J. Gómez-García and J. M. Martínez-Agudo, Adv. Mater., 1999, 11, 558 CrossRef CAS; (c) E. Coronado, J. R. Galán-Mascarós, C. J. Gómez-García, J. Ensling and P. Gutlich, Chem.–Eur. J., 2000, 6, 552 CrossRef CAS.
- (a) S. Bénard, P. Yu, J. P. Audière, E. Rivière, R. Clèment, J. Ghilhem, L. Tchertanov and K. Nakatami, J. Am. Chem. Soc., 2000, 122, 9444 CrossRef CAS; (b) S. M. Aldoshin, N. A. Sanina, V. I. Minkin, N. A. Voloshin, V. N. Ikorskii, V. I. Ovcharenko, V. A. Smirnov and N. K. Nagaeva, J. Mol. Struct., 2007, 826, 6 CrossRef CAS.
- (a) E. Coronado, J. R. Galán-Mascarós, C. J. Gómez-García and V. Laukhin, Nature, 2000, 408, 447 CrossRef CAS; (b) A. Alberola, E. Coronado, J. R. Galán-Mascarós, C. Giménez-Saiz and C. J. Gómez-García, J. Am. Chem. Soc., 2003, 125, 10774 CrossRef CAS; (c) E. Coronado, J. R. Galán-Mascarós, C. J. Gómez-García, E. Martínez-Ferrero and S. Van Smaalen, Inorg. Chem., 2004, 43, 4808 CrossRef CAS.
- H. Okawa, A. Shigematsu, M. Sadakiyo, T. Miyagawa, K. Yoneda, M. Ohba and H. Kitagawa, J. Am. Chem. Soc., 2009, 131, 13516 CrossRef CAS.
- (a) R. Andrés, M. Gruselle, B. Malézieux, M. Verdaguer and J. Vaissermann, Inorg. Chem., 1999, 38, 4637 CrossRef CAS; (b) R. Andrés, M. Brissard, M. Gruselle, C. Train, J. Vaissermann, B. Malézieux, J. P. Jamet and M. Verdaguer, Inorg. Chem., 2001, 40, 4633 CrossRef CAS; (c) M. Clemente-León, E. Coronado, J. C. Dias, A. Soriano-Portillo and R. D. Willett, Inorg. Chem., 2008, 47, 6458 CrossRef CAS.
- J. R. Galán-Mascarós, E. Coronado, P. A. Goddard, J. Singleton, A. I. Coldea, J. D. Wallis, S. J. Coles and A. Alberola, J. Am. Chem. Soc., 2010, 132, 9271 CrossRef CAS.
- (a) C. Train, R. Gheorghe, V. Krstic, L. M. Chamoreau, N. S. Ovanesyan, G. L. J. A. Rikken, M. Gruselle and M. Verdaguer, Nat. Mater., 2008, 17, 729 CrossRef; (b) C. Train, T. Nuida, R. Gheorghe, M. Gruselle and S. Ohkoshi, J. Am. Chem. Soc., 2009, 131, 16838 CrossRef CAS.
- (a) R. Sieber, S. Decurtins, H. Stoeckli-Evans, C. Wilson, D. Yufit, J. A. K. Howard, S. C. Capelli and A. Hauser, Chem.–Eur. J., 2000, 6, 361 CrossRef CAS; (b) E. Coronado, J. R. Galán-Mascarós, M. C. Giménez-López, M. Almeida and J. C. Waerenborgh, Polyhedron, 2007, 26, 1838 CrossRef CAS; (c) M. Clemente-León, E. Coronado, M. C. Giménez-López, A. Soriano-Portillo, J. C. Waerenborgh, F. S. Delgado and C. Ruiz-Pérez, Inorg. Chem., 2008, 47, 9111 CrossRef CAS; (d) M. Clemente-León, E. Coronado and M. López-Jordà, Dalton Trans., 2010, 39, 4903 RSC.
- M. Clemente-León, E. Coronado, M. López-Jordà, G. Mínguez Espallargas, A. Soriano-Portillo and J. C. Waerenborgh, Chem.–Eur. J., 2010, 16, 2207 CrossRef CAS.
- S. Decurtins, P. Gütlich, K. M. Hasselbach, A. Hauser and H. Spiering, Inorg. Chem., 1985, 24, 2174 CrossRef CAS.
- C. Enachescu, A. Hauser, J.-J. Girerd and M.-L. Boillot, ChemPhysChem, 2006, 7, 1127 CrossRef CAS.
- (a) S. Hayami, Z.-Z. Gu, M. Shiro, Y. Einaga, A. Fujishima and O. Sato, J. Am. Chem. Soc., 2000, 122, 7126 CrossRef CAS; (b) G. Juhász, S. Hayami, O. Sato and Y. Maeda, Chem. Phys. Lett., 2002, 364, 164 CrossRef CAS.
- S. Hayami, K. Hiki, T. Kawahara, Y. Maeda, D. Urakami, K. Inoue, M. Ohama, S. Kawata and O. Sato, Chem.–Eur. J., 2009, 15, 3497 CrossRef CAS.
- Crystal data for 1 at 300 K: C27H26Cl2CrFeMnN4O14, M = 864.21, monoclinic, space groupP21/c (no. 14), a = 12.096(5) Å;, b = 32.135(5) Å;, c = 9.467(5) Å;, β = 111.981(5)°, V = 3412(2) Å3, Z = 4, μ = 1.325 mm−1, F(000) = 1748, T = 300(2)K, 44282 reflections measured, 11352 unique (Rint = 0.0589), R1 = 0.0635, wR2 = 0.1689 for I > 2σ(I). CCDC 807173. Crystal data for 1 at 250 K: C27H26Cl2CrFeMnN4O14, M = 864.21, monoclinic, space groupP21/c (no. 14), a = 11.9215(15) Å, b = 31.910(3) Å, c = 9.4841(11) Å, β = 111.535(13)°, V = 3356.1(6) Å3, Z = 4, μ = 1.348 mm−1, F(000) = 1748, T = 250(2)K, 43973 reflections measured, 11354 unique (Rint = 0.0590), R1 = 0.0530, wR2 = 0.1240 for I > 2σ(I). CCDC 813345. Crystal data for 1 at 120 K: C27H26Cl2CrFeMnN4O14, M = 864.21, monoclinic, space groupP21/c (no. 14), a = 11.5561(3) Å, b = 31.6369(7) Å, c = 9.5457(3) Å, β = 109.984(3)°, V = 3279.77(15) Å3, Z = 4, μ = 1.379 mm−1, F(000) = 1748, T = 120(2)K, 26791 reflections measured, 10705 unique (Rint = 0.0302), R1 = 0.0477, wR2 = 0.0951 for I > 2σ(I). CCDC 807172. Crystal data for 1 at 92 K: C27H26Cl2CrFeMnN4O14, M = 864.21, monoclinic, space groupP21/c (no. 14), a = 11.5602(4) Å;, b = 31.6268(12) Å;, c = 9.5237(4) Å;, β = 110.199(4)°, V = 3267.8(2) Å3, Z = 4, μ = 1.379 mm−1, F(000) = 1748, T = 92(2)K, 29715 reflections measured, 10846 unique (Rint = 0.0557), R1 = 0.0546, wR2 = 0.1230 for I > 2σ(I). CCDC 813714. Crystal data of 1 at 180 K have been taken from ref. 14.
- P. Guionneau, M. Marchivie, G. Bravic, J.-F. Létard and D. Chasseau, Top. Curr. Chem., 2004, 234, 97 CAS.
- J. K. McCusker, A. L. Rheingold and D. N. Hendrickson, Inorg. Chem., 1996, 35, 2100 CrossRef CAS.
- M. Marchivie, P. Guionneau, J.-F. Létard and D. Chasseau, Acta Crystallogr., Sect. B: Struct. Sci., 2005, 61, 25 CrossRef.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885 RSC.
- L. Néel, Ann. Phys., 1948, 3, 137 CAS.
- J.-F. Létard, J. Mater. Chem., 2006, 16, 2550 RSC.
- A. Hauser, J. Adler and P. Gütlich, Chem. Phys. Lett., 1988, 152, 468 CrossRef CAS.
- A. Hauser, J. Chem. Phys., 1991, 94, 2741 CrossRef CAS.
- A. Hauser, Top. Curr. Chem., 2004, 234, 155 CAS.
- S. Schenker, A. Hauser and R. M. Dyson, Inorg. Chem., 1996, 35, 4676 CrossRef CAS.
- S. Dorbes, L. Valade, J. A. Real and C. Faulmann, Chem. Commun., 2005, 69 RSC.
- R. Pritchard, S. A. Barrett, C. A. Kilner and M. A. Halcrow, Dalton Trans., 2008, 3159 RSC.
- C. Faulmann, P. A. Szilágyi, K. Jacob, J. Chahine and L. Valade, New J. Chem., 2009, 33, 1268 RSC.
- V. Mishra, R. Mukherjee, J. Linares, C. Balde, C. Desplanches, J.-F. Létard, E. Collet, L. Toupet, M. Castro and F. Varret, Inorg. Chem., 2008, 47, 7577 CrossRef CAS.
- C. Faulmann, S. Dorbes, J. A. Real and L. Valade, J. Low Temp. Phys., 2006, 142, 261 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: crystallographic data, intermolecular interactions, ZFC and FC magnetisation and AC measurements before and after irradiation, and details of the fitting of the magnetism of [MnIICrIII(ox)3] layers. CCDC reference numbers 807172, 807173, 813345, 813714. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00015b |
| This journal is © The Royal Society of Chemistry 2011 |