Cylindrical micelles from the living crystallization-driven self-assembly of poly(lactide)-containing block copolymers†
Nikos
Petzetakis
,
Andrew P.
Dove
* and
Rachel K.
O'Reilly
*
Department of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: a.p.dove@warwick.ac.uk; r.k.o-reilly@warwick.ac.uk
First published on 15th February 2011
Abstract
The synthesis and self-assembly of poly(lactide)-b-poly(acrylic acid) and poly(lactide)-b-poly(dimethylaminoethylacrylate) block copolymers by a combination of ring-opening polymerization and reverse-addition fragmentation chain transfer (RAFT) polymerization is reported. Self-assembly of block copolymers containing enantiopure homochiral poly(lactide), PLA, by a simple direct dissolution methodology results in core-crystallization to afford micelles with a cylindrical morphology. Amorphous atactic PLA cores and conditions that did not promote crystallization resulted in spherical micelles. Cylindrical micelles were characterized by transmission electron microscopy (TEM) with cryo-TEM, small angle neutron scattering (SANS) and angular dependent dynamic light scattering (DLS) proving that the cylindrical morphology was persistent in solution. Manipulation of the assembly conditions enabled the length and dispersity of the resultant cylindrical micelles to be controlled.
Introduction
The shape of self-assembled nanoparticles can play an important role in their application. Cylindrical particles, for example, are realizing advantages in comparison to spherical micellar morphologies with preferable properties being elicited in their application as templates for spherical micelles and nanowires as well as toughening of polymer resins.1 Perhaps the most notable is their application in the biomedical arena, with cylindrical micelles reported to display increased in vivo circulation times and altered cell internalization pathways compared to analogous spherical morphologies.2–5 Understanding and directing the forces that determine the morphological control in the micellar self-assembly process is a key factor in fully realizing this potential.6–8While several synthetic methods have been utilized in the synthesis of cylindrical micelles,9–12 facile and versatile synthetic methodologies are essential to enable advances in the field. The synthesis of unimolecular micelles/bottle brush copolymers has been shown to yield core-shell cylindrical nanoparticles.11,13,14 This approach enables exquisite control over the molecular parameters, however often involves lengthy synthetic procedures. Harnessing the hydrophobic effect within a narrow window to realize worm-like micelles has also been highly successful for a range of copolymers.12,15–18 However, the self-assembly process can be greatly affected by a range of parameters including assembly conditions,7 mid-block interactions,19 electrostatics and crystallization.6,8 This latter phenomenon has been exploited to great effect by Manners, Winnik and coworkers in the synthesis of poly(ferrocenyl silane) block copolymers to generate nanoscopic cylinders with controlled geometries.8,20–26 Given the considerable potential to utilize shape-specific micelles and particles, we sought to synthesize biorelevant cylindrical block copolymer micelles with highly controlled geometries via a simple self assembly methodology that would result in particles with a wide range of possible modifications to enable stabilization by shell crosslinking, hollowing by core degradation and careful positioning of a range of functional groups.
To this end, our studies focus on the synthesis and application of poly(lactide)-containing block copolymers. Poly(lactide), PLA, is a biodegradable and biocompatible polymer that as either homochiral enantiomer is semi-crystalline. PLA has been studied for many biomedical applications and is widely approved for in vivo applications.27–29 Several previous studies have investigated the effects of semi-crystalline cores (such as poly(butadiene), poly(ethylene oxide) and poly(caprolactone)) and have demonstrated that a wide range of structures have been identified, including cylinders,30–33 however to date PLA-only cores have not been manipulated in this manner despite the obvious material advantages of PLA. While several reports detail the self-assembly of the PLA-stereocomplex (an equimolar mixture of L and D-PLA that leads to increased crystallinity with respect to either homopolymer) to afford spherical micelles,34–38 a range of unexpected geometries including onion-like and helical micelles have also been observed.39–42 Notably, Bouteiller and coworkers have utilized PLA stereocomplexation to drive the formation of anisotropic cylindrical PLA-b-poly(caprolactone) block copolymer aggregates in THF.43 We postulated that manipulation of the self-assembly conditions to enable more efficient crystallization of a homochiral PLA core would result in the isolation of cylindrical micelle structures. Specifically that self-assembly of PLA-containing amphiphilic block copolymers above the glass transition temperature of the PLA (typically ca. 55–60 °C)44 segment would allow the chains to orient within the core of the micelles, thus facilitating crystallization, influencing the self assembly process to override the hydrophobic effect and alter the resultant micelle morphology in a simple procedure.
Results and discussion
Synthesis of initial PLA-poly(acrylic acid) block copolymers was targeted using a combination of ring-opening polymerization (ROP) and reverse-addition fragmentation chain transfer (RAFT) polymerization techniques from a dual-headed initiator/chain transfer agent, 1, to allow facile synthesis of a range of block copolymers (Scheme 1).45Polymers were targeted with large hydrophilic fractions (>70% by weight) such that they would be expected to yield spherical micelles under standard solution assembly conditions. | ||
| Scheme 1 Preparation of copolymers2–4. Conditions: i) Lactide, 1-(3,5-bis(trifluoromethyl)phenyl)-3-cyclohexyl-thiourea, (-)-sparteine, CH2Cl2; ii) AIBN, CHCl3, THP-acrylate; iii) AcOH, THF–H2O at 65 °C. | ||
The ROP of lactide was undertaken in dichloromethane, mediated by a highly selective thiourea/tertiary amine catalyst system as reported previously.46 In this manner, trithiocarbonate end-functional poly(L-LA)32, poly(D-LA)42 and atactic poly(DL-LA)33 were prepared by the ROP of L-LA, D-LA and rac-LA, respectively. Initial attempts to grow a second polymer block from the PLA macro-CTA via RAFT were unsuccessful in anisole, DMF, DMSO and dioxane, resulting in bimodal traces when detected by GPC UV analysis at 309 nm (corresponding to λmax of 1). However, the PLA polymers could be successfully utilized as macro-CTAs in RAFT polymerization by using chloroform as a solvent (Table 1). The poly(acrylic acid), PAA, block was synthesized by deprotection following the radical-initiated RAFT polymerization of a THP-acrylate monomer (Scheme 1). 1H NMR and GPC analysis (using RI and UV detectors) indicated that the chain growth had occurred cleanly and without any adverse affect on the PLA block. The PLA-b-PAA copolymers (2–4) were realized by deprotection of the THP groups under mild acidic conditions with subsequent analysis of the 1H NMR spectra (loss of signal at ca. δ 6.0) and IR analysis (loss of carbonyl absorbance at 1736 cm−1). Furthermore, analysis of the homonuclear decoupled 1H NMR spectra of the PLA macro-CTA and PLA-b-PAA diblock copolymer in d6-DMSO displayed no discernable changes indicating that no degradation or epimerization of the PLA blocks had occurred upon deprotection (see SI†). To further confirm the absence of degradation, a sample of PLA homopolymer was subjected to the same conditions and again analysis by 1H NMR and GPC indicated that no adverse reactions were occurring.
| M n NMR a (kDa) | M n GPC b (kDa) | M w/Mnb | DP(PLA) a | DP(PTHPA or PDMAEMA)a | |
|---|---|---|---|---|---|
| a Determined by 1H NMR spectroscopy. b Determined by GPC analysis in THF. c Determined by GPC analysis in DMF. | |||||
| P(L-LA) | 5.0 | 9.4 | 1.08 | 32 | — |
| P(D-LA) | 6.5 | 12.4 | 1.05 | 43 | — |
| P(DL-LA) | 4.6 | 8.4 | 1.06 | 33 | — |
| PTHPA-b-P(L-LA) | 46.3 | 65.0 | 1.42 | 32 | 265 |
| PTHPA-b-P(D-LA) | 34.4 | 54.1 | 1.38 | 42 | 220 |
| PTHPA-b-P(DL-LA) | 37.4 | 59.7 | 1.31 | 33 | 240 |
| PDMAEA-b-P(L-LA), 8 | 27.2 | 12.9c | 1.48c | 32 | 190 |
Initially the self assembly of the three diblock copolymers (2–4) was explored by direct dissolution in nanopure H2O with a polymer concentration of 0.25 mg mL−1 at ambient temperature with overnight stirring. In all three cases, dynamic light scattering (DLS) analysis revealed that large ill-defined aggregates (PD's > 0.3) were formed. To further explore the assembly of these amphiphilic diblocks, the solutions were heated to increase the assembly temperature above the Tg of the PLA to aid dissolution and facilitate crystallization of the core block during the assembly process. Following dissolution of the block copolymers at 65 °C for 1 h followed by rapid cooling, DLS analysis revealed the formation of large well-defined aggregates (PDsca. 0.1 - Table 2). These assemblies were further characterized by TEM analysis using phosphotungstic acid as a stain (Fig. 1). Notably, the assemblies formed from either enantiopure homochiral PLA core blocks (2 and 3) were cylindrical in morphology (with average lengths, Dl (5) = 213 nm and Dl (6) = 179 nm) whereas those containing an atactic PLA core block, that was not able to crystallize (4), afforded spherical structures (Dav(7) = 80 nm).
| D h,app DLS (nm) | D l TEM (nm) | L w/Ln | ||||
|---|---|---|---|---|---|---|
| Vol. | Num. | Int. | PD | |||
| PAA-b-P(L-LA), 5 | 185.5 | 158.7 | 186.5 | 0.120 | 213 | 1.08 |
| PAA-b-P(D-LA), 6 | 175.7 | 146.0 | 178.6 | 0.080 | 179 | 1.08 |
| PAA-b-P(DL-LA), 7 | 102.3 | 80.70 | 119.7 | 0.156 | 21 | — |
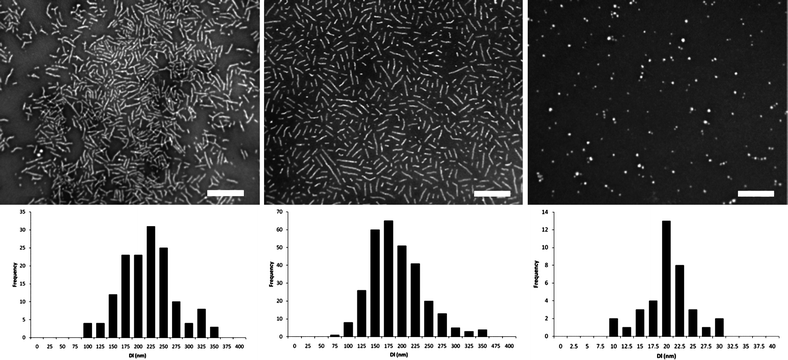 | ||
| Fig. 1 TEM data for (from left to right) enantiomeric (5 and 6) and racemic (7) assemblies formed by self assembly at 65 °C. Scale bar = 500 nm. | ||
To further probe these observations and confirm that these morphologies were present in solution, angular dependent light scattering measurements were performed for cylindrical assembly, 5, formed from the poly(L-LA) diblock, 2. From these measurements it was clear that the diffusion coefficient depended on the scattering angle, further indicating a non-spherical solution morphology for 5.47,48 Further data obtained by Small Angle Neutron Scattering (SANS) analysis for micelle 6 at 1 mg mL−1 in D2O confirmed a non-spherical solution morphology which could be best fitted to a cylindrical model, with a length of >150 nm and a diameter of ca. 15 nm, which correlates well with previous observations (see SI for further discussion of the fitting for this system†). In addition, both the poly(L-LA)-PAA assembly (5) and poly(DL-LA)-PAA assembly (7) were analyzed by cryo-TEM (using a cryo plunger and double blot technique). In correlation with the solution and TEM studies, the structures containing the semi-crystalline poly(L-LA) were observed as cylinders, contrasting the spherical assemblies obtained under identical conditions using the amorphous atactic poly(DL-LA)-containing assemblies (Dl (5) = 170 nm and Dav (7) = 100 nm) (Fig. 2). Using a combination of the cryo-TEM images with positively stained TEM (see SI†) images the cylinder corona could be estimated to be around 4–5 ± 1nm.
 | ||
| Fig. 2 Cryo-TEM for enantiomeric (5) (RHS) and racemic (7) assemblies (LHS) formed by self assembly at 65 °C. Scale bar = 200 nm. Note that in the RHS image film formation is also observed at the hole edge and we propose this is an artifact of the cryo-TEM analysis. | ||
To probe the semi-crystalline nature of the homochiral PLA cores, Wide-Angle X-Ray Diffraction (WAXD) studies on micelles 5 and 7 were undertaken. Analysis of a sample of micelles 5 prepared by freeze drying prior to deposition as a thin film on a silica wafer revealed a slightly broadened peak at a 2θ angle of 16.6°, consistent with that expected for the strong (110)/(200) reflection in the crystalline domains of poly(L-LA) (Fig. 3a).49 Notably, identical analysis of a sample of micelles 7 did not reveal any strong reflections, only a broad peak (Fig. 3b). These data clearly demonstrate a difference in the crystallinity between the micelles, which strongly indicates that the presence of crystallinity within the core PLA domain is essential for the formation of cylindrical micelles. Additionally, in order to demonstrate that these effects were not a consequence of the use of a PAA corona, we also prepared a second amphiphilic block with a poly(dimethylaminoethylacrylate) hydrophilic block. Copolymer8 (Table 1; PDMAEA190-b-P(L-LA)32) was prepared by a similar method to that reported for the initial blocks and was self assembled under identical conditions. Once again cylindrical micelles (Dl (9) = 130 nm; Table 2) were observed by TEM (see SI†). It should be noted that the micelles formed from all polymers utilized in this study were stable for extended periods of time (>6 months) under normal benchtop conditions.
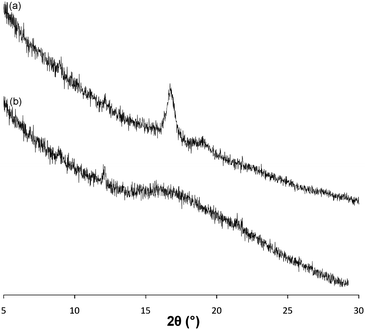 | ||
| Fig. 3 WAXD patterns for (a) micelles 5 and (b) micelles 7. | ||
To further investigate the assembly and confirm the importance of the crystallization process, the self assembly of the P(L-LA)-b-PAA block copolymer (2) was further examined under a range of conditions. In order to confirm that the assembly temperature had a key role in the formation of cylindrical structures through crystallization affects rather than improved solubility the polymer, 2 (PAA-b-P(L-LA)) was also assembled using conventional solvent switching methods (Acetone-Water) as reported by LeRoux and coworkers.37 Using this method, relatively well defined spherical particles were observed by DLS (Dav = 118 nm, PD = 0.108) and TEM analysis (Dav = 99 nm) (Fig. 4).
 | ||
| Fig. 4 TEM image of micelles 9 prepared from polymer2 by a solvent switch approach (scale bar = 100 nm). | ||
With the requirement for conditions that promote effective crystallization established, we sought to further investigate the effect of time and dissolution temperature on the micelles formed. Maintaining a heating time of 1 h but lowering the assembly temperature to 25 °C led to the observation of large ill-defined but largely spherical aggregates, while dissolution of the block copolymer at 95 °C for either 20 mins or 1 h led to the observation of largely cylindrical structures, although less well defined cylinders and notable aggregation was observed (Fig. 5). Such studies indicate that the observed cylindrical structures are kinetically trapped stable intermediates that can only be accessed through core-crystallization above the Tg of the PLA block.
 | ||
| Fig. 5 TEM data for the self-assembly of polymer3 for 1 h at (a) 25 °C; (b) 60 °C and (c) 95 °C, scale bar = 300 nm. | ||
Investigation of the effect of heating times on the formation of the cylindrical micelles revealed that, in common with the preparation of micelles at higher temperatures, large poorly defined structures were observed at extended periods (see SI†). Reducing the heating times that the polymer solution was held at 65 °C however, led to the observation of cylinders of reduced length, determined by both DLS and TEM images (Fig. 6). Furthermore, a plot of the average length of the cylinders (Dl) against the dissolution time observed a linear relationship (Fig. 7). These data suggest a linear growth of the cylindrical micelles such that the length of the cylinders can be kinetically controlled. These observations mirror the living epitaxial micelle growth observed by Manners, Winnik and coworkers, showing that these principles are applicable to organic polymer cores thus expanding the scope of possible polymer systems with which to synthesize cylindrical micelles of highly controlled size and functionality.21,23,24
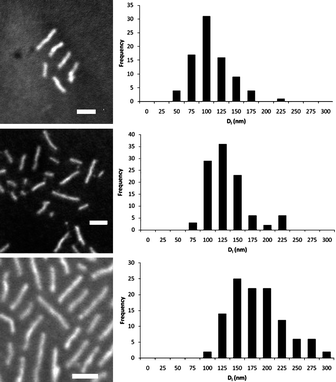 | ||
| Fig. 6 TEM images and histograms for cylindrical micelles prepared from self assembly of PAA-b-P(L-LA), 2, at 65 °C in H2O, 0.25 mg mL−1, a) 10 min, b) 20 mins and c) 40 mins. Scale bar = 100 nm. | ||
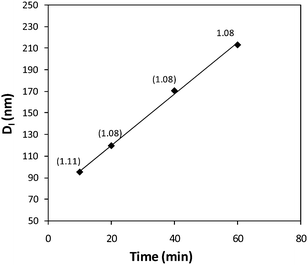 | ||
| Fig. 7 Linear correlation between average micelle length (Dl, determined by TEM analysis) and assembly time for the self assembly of PAA-b-P(L-LA), 2, at 65 °C in H2O, 0.25 mg mL−1. | ||
Further evidence of the living growth of the cylindrical micelles was obtained by the investigation of reinitiation of cylinder growth, using a similar polymer2a (PAA300-b-P(L-LA)32) to afford micelles 5a. To this end, a solution of 5a was heated in nanopure water at 65 °C (25 mg mL−1) for 10 mins. DLS and TEM analysis of the micelles formed was consistent with that previously observed for the related polymer2 (Dl = 144 nm). These micelles were stable for extended periods of time at room temperature. However, reheating this solution to 65 °C for a further 50 min led to reinitiation of cylinder growth such that the cylinders had similar dimensions to that reported without stopping the growth at 10 min (Dav = 248 nm, PD = 0.232; Dl = 200 nm, Lw/Ln = 1.06 compared to Dl = 202 nm, Lw/Ln = 1.08) (see SI†). These observations are entirely consistent with an epitaxial growth process of the cylinders by the addition of remaining unimers following an initial nucleation event. Clearly, this initial event is critical to both the final dimensions but also the dispersity of the resultant micellar assemblies.
Polymer 2a (PAA300-b-P(L-LA)32) showed very similar living growth kinetics to the original polymer2 with both systems affording initial micelle sizes after 10 min of around 100 nm and final sizes after 60 mins ca. 210 nm. To explore the effect of polymer concentration on the nucleation event and also kinetics of micelle growth, the micellization of 2a was further explored at a higher polymer concentration (0.5 mg mL−1) at 65 °C. After 10 min of heating, both systems showed cylinders of different lengths by TEM, with the higher concentration solution producing cylinders of longer initial lengths (Dl 144 nm versus 100 nm). Both concentrations showed a linear increase in cylinder size with increasing assembly time at 65 °C as observed in our initial system and after 60 min the solution with higher polymer concentration had afforded slightly longer cylindrical structures (232 versus 202 nm). Crucially the rate of growth is comparable at both concentrations which indicates that changing the initial polymer concentration affects the nucleation event but not the living growth process (Fig. 8). Furthermore initial investigations indicate that the cooling rate does not affect the size or dispersity of the resultant structures. Interestingly it seems that for increased heating times a leveling off of the micelle size is observed in both cases, suggesting that there is an optimum window for the controlled self assembly of cylindrical micelles in this system.
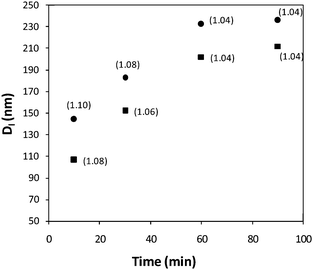 | ||
| Fig. 8 Plot of average micelle length (Dl, determined by TEM analysis) and assembly time with Lw/Ln in parenthesis, for the self assembly of PAA-b-P(L-LA), 2a, at 65 °C in H2O, 0.25 mg mL−1 (■) and 0.5 mg mL−1 (●). | ||
Conclusions
This work demonstrates the living crystallization-driven self-assembly of readily accessible PLA-containing block copolymers through a thermally controlled crystallization. The anisotropic nature of the micelles has been characterized by a range of analytical techniques with all data consistent with the formation of cylinders in solution. The simple assembly procedures reported herein, in combination with the application of accessible RAFT and ROP synthetic methodologies, enables unprecedented versatility in the preparation of well-defined cylindrical micelles. Furthermore the living epitaxial growth of these cylinders has been demonstrated thus enabling the preparation of cylinders of controlled dimensions.Acknowledgements
We gratefully acknowledge the support provided by EPSRC (EP/G004897/1) for funding a fellowship (R.O'R) and a studentship for N.P. The Research Councils UK (RCUK) are also acknowledged for funding a fellowship to A.P.D. Some of the equipment used in this research was obtained through Birmingham Science City with support from Advantage West Midlands (AWM) and the European Regional Development Fund (ERDF). Purac are thanked for their generous donation of lactide. Drs Steve Furzeland and Derek Atkins (Unilever) are thanked for cryo-TEM analysis, Dr Sarah Rogers (ISIS, Rutherford Appleton Laboratories) and STFC are acknowledged for SANS Xpress beamtime at ISIS for SANS analysis and Dr David Walker, Daniel Baker and Prof. Pam Thomas (Department of Physics, University of Warwick) for WAXD analysis.Notes and references
- J. Liu, Z. J. Thompson, H.-J. Sue, F. S. Bates, M. A. Hillmyer, M. Dettloff, G. Jacob, N. Verghese and H. Pham, Macromolecules, 2010, 43, 7238–7243 CrossRef CAS.
- Y. Geng, P. Dalhaimer, S. S. Cai, R. Tsai, M. Tewari, T. Minko and D. E. Discher, Nat. Nanotechnol., 2007, 2, 249–255 CrossRef CAS.
- K. Zhang, H. F. Fang, Z. Y. Chen, J. S. A. Taylor and K. L. Wooley, Bioconjugate Chem., 2008, 19, 1880–1887 CrossRef CAS.
- K. Zhang, R. Rossin, A. Hagooly, Z. Y. Chen, M. J. Welch and K. L. Wooley, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7578–7583 CrossRef CAS.
- A. Harada and K. Kataoka, Prog. Polym. Sci., 2006, 31, 949–982 CrossRef CAS.
- F. Grohn, Macromol. Chem. Phys., 2008, 209, 2295–2301 CrossRef.
- R. C. Hayward and D. J. Pochan, Macromolecules, 2010, 43, 3577–3584 CrossRef CAS.
- M. Lazzari and M. A. Lopez-Quintela, Macromol. Rapid Commun., 2009, 30, 1785–1791 CrossRef CAS.
- H. Ajiro and M. Akashi, Macromol. Rapid Commun., 2010, 31, 714–717 CrossRef CAS.
- Q. J. Chen, H. Zhao, T. Ming, J. F. Wang and C. Wu, J. Am. Chem. Soc., 2009, 131, 16650–16651 CrossRef CAS.
- C. Cheng, K. Qi, D. S. Germack, E. Khoshdel and K. L. Wooley, Adv. Mater., 2007, 19, 2830–2835 CrossRef CAS.
- C. A. Dreiss, Soft Matter, 2007, 3, 956–970 RSC.
- K. Huang, D. P. Canterbury and J. Rzayev, Macromolecules, 2010, 43, 6632–6638 CrossRef CAS.
- K. Huang and J. Rzayev, J. Am. Chem. Soc., 2009, 131, 6880–6885 CrossRef CAS.
- J. Grumelard, A. Taubert and W. Meier, Chem. Commun., 2004, 1462–1463 RSC.
- I. W. Hamley, Soft Matter, 2005, 1, 36–43 RSC.
- Y. Y. Won, H. T. Davis and F. S. Bates, Science, 1999, 283, 960–963 CrossRef CAS.
- L. Zhang and A. Eisenberg, Science, 1995, 268, 1728–1731 CrossRef CAS.
- S. H. Kim, F. Nederberg, R. Jakobs, J. P. K. Tan, K. Fukushima, A. Nelson, E. W. Meijer, Y. Y. Yang and J. L. Hedrick, Angew. Chem., Int. Ed., 2009, 48, 4508–4512 CrossRef CAS.
- J. B. Gilroy, T. Gädt, G. R. Whittell, L. Chabanne, J. M. Mitchels, R. M. Richardson, M. A. Winnik and I. Manners, Nat. Chem., 2010, 2, 566–570 CrossRef CAS.
- T. Gadt, N. S. Ieong, G. Cambridge, M. A. Winnik and I. Manners, Nat. Mater., 2009, 8, 144–150 CrossRef.
- D. A. Rider and I. Manners, Polym. Rev., 2007, 47, 165–195 Search PubMed.
- H. Wang, W. J. Lin, K. P. Fritz, G. D. Scholes, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2007, 129, 12924–12925 CrossRef CAS.
- X. S. Wang, G. Guerin, H. Wang, Y. S. Wang, I. Manners and M. A. Winnik, Science, 2007, 317, 644–647 CrossRef CAS.
- J. Massey, K. N. Power, I. Manners and M. A. Winnik, J. Am. Chem. Soc., 1998, 120, 9533–9540 CrossRef CAS.
- J. A. Massey, K. Temple, L. Cao, Y. Rharbi, J. Raez, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2000, 122, 11577–11584 CrossRef CAS.
- A. C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466–1486 CrossRef CAS.
- E. S. Place, J. H. George, C. K. Williams and M. M. Stevens, Chem. Soc. Rev., 2009, 38, 1139–1151 RSC.
- K. E. Uhrich, S. M. Cannizzaro, R. S. Langer and K. M. Shakesheff, Chem. Rev., 1999, 99, 3181–3198 CrossRef CAS.
- A. M. Mihut, M. Drechsler, M. Moller and M. Ballauff, Macromol. Rapid Commun., 2010, 31, 449–453 CrossRef CAS.
- C.-K. Chen, S.-C. Lin, R.-M. Ho, Y.-W. Chiang and B. Lotz, Macromolecules, 2010, 43, 7752–7758 CrossRef CAS.
- J. Zhang, L. Q. Wang, H. J. Wang and K. H. Tu, Biomacromolecules, 2006, 7, 2492–2500 CrossRef CAS.
- T. Vilgis and A. Halperin, Macromolecules, 1991, 24, 2090–2095 CrossRef CAS.
- K. Fukushima, R. C. Pratt, F. Nederberg, J. P. K. Tan, Y. Y. Yang, R. M. Waymouth and J. L. Hedrick, Biomacromolecules, 2008, 9, 3051–3056 CrossRef CAS.
- A. Bishara, H. R. Kricheldorf and A. J. Domb, Macromol. Symp., 2005, 225, 17–30 CrossRef CAS.
- L. Chen, Z. G. Xie, J. L. Hu, X. S. Chen and X. B. Jing, J. Nanopart. Res., 2007, 9, 777–785 CrossRef CAS.
- N. Kang, M. E. Perron, R. E. Prud'homme, Y. B. Zhang, G. Gaucher and J. C. Leroux, Nano Lett., 2005, 5, 315–319 CrossRef CAS.
- S. H. Kim, J. P. K. Tan, F. Nederberg, K. Fukushima, Y. Y. Yang, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2009, 42, 25–29 CrossRef CAS.
- R. M. Ho, C. K. Chen, Y. W. Chiang, B. T. Ko and C. C. Lin, Adv. Mater., 2006, 18, 2355–2358 CrossRef CAS.
- J. L. Hu, Z. H. Tang, X. Y. Qiu, X. Pang, Y. K. Yang, X. S. Chen and X. B. Jing, Biomacromolecules, 2005, 6, 2843–2850 CrossRef CAS.
- L. Sun, J. E. Ginorio, L. Zhu, I. Sics, L. X. Rong and B. S. Hsiao, Macromolecules, 2006, 39, 8203–8206 CrossRef CAS.
- L. Sun, L. Zhu, L. X. Rong and B. S. Hsiao, Angew. Chem., Int. Ed., 2006, 45, 7373–7376 CrossRef CAS.
- D. Portinha, F. Boue, L. Bouteiller, G. Carrot, C. Chassenieux, S. Pensec and G. Reiter, Macromolecules, 2007, 40, 4037–4042 CrossRef CAS.
- J. M. Becker, R. J. Pounder and A. P. Dove, Macromol. Rapid Commun., 2010, 31, 1923–1937 CrossRef CAS.
- J. Skey and R. K. O'Reilly, Chem. Commun., 2008, 4183–4185 RSC.
- R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, P. N. P. Lundberg, A. P. Dove, H. B. Li, C. G. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 7863–7871 CrossRef CAS.
- G. Guerin, J. Raez, I. Manners and M. A. Winnik, Macromolecules, 2005, 38, 7819–7827 CrossRef CAS.
- W. Burchard, Adv. Polym. Sci., 1983, 48, 1–124 CAS.
- P. Pan, W. Kai, B. Zhu, T. Dong and Y. Inoue, Macromolecules, 2007, 40, 6898–6905 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and further characterization of polymers and assemblies. See DOI: 10.1039/c0sc00596g |
| This journal is © The Royal Society of Chemistry 2011 |