Synthesis and utility of fluorogenic acetoxymethyl ethers†
Luke D.
Lavis
ab,
Tzu-Yuan
Chao
c and
Ronald T.
Raines
*bc
aJanelia Farm Research Campus, Howard Hughes Medical Institute, 19700 Helix Drive, Ashburn, VA 20147, USA
bDepartment of Chemistry, University of Wisconsin–Madison, 1101 University Avenue, Madison, WI 53706, USA
cDepartment of Biochemistry, University of Wisconsin–Madison, 433 Babcock Drive, Madison, WI 53706, USA. E-mail: rtraines@wisc.edu
First published on 15th November 2010
Abstract
Phenolic fluorophores such as fluorescein, Tokyo Green, resorufin, and their derivatives are workhorses of biological science. Acylating the phenolic hydroxyl group(s) in these fluorophores masks their fluorescence. The ensuing ester is a substrate for cellular esterases, which can restore fluorescence. These esters are, however, notoriously unstable to hydrolysis, severely compromising their utility. The acetoxymethyl (AM) group is an esterase-sensitive motif that can mask polar functionalities in small molecules. Here, we report on the use of AM ether groups to mask phenolic fluorophores. The resulting profluorophores have a desirable combination of low background fluorescence, high chemical stability, and high enzymatic reactivity, both in vitro and in cellulo. These simple phenyl ether-based profluorophores could supplement or supplant the use of phenyl esters for imaging biochemical and biological systems.
Introduction
The utility of small-molecule fluorescent probes is well established in biology.1 In particular, fluorogenic esterase substrates are vital tools for examining biological and biochemical processes. Such compounds can be used to assess cell viability,2 to measure enzymatic activity,3 as fluorogenic labels for biomolecular imaging,4 or as part of a delivery strategy for fluorescent ion indicators.5,6 Chemical “masking” of polar functionalities with ester groups can stifle fluorescence and facilitate efficient diffusion across lipid bilayers.7,8 Esterases endogenous in eukaryotic cells are able to catalyze the hydrolysis of some synthetic ester bonds,9 thereby releasing the fluorescent molecule.The archetype of fluorogenic esterase substrates is based on the antique fluorophore fluorescein (1a; Scheme 1).10 Functionalization of the phenolic oxygens found in fluorescein with a suitable acylating agent (e.g., acetic anhydride) gives fluorescein diacetate (FDA; 2a). This acyl substitution secures the molecule in a colorless, nonfluorescent lactone form. Rotman and Papermaster demonstrated that the lipophilic FDA can cross cell membranes and be activated by cellular esterases.7 This diester strategy has been used to mask numerous fluorescein derivatives for intracellular delivery.2
 | ||
| Scheme 1 Fluorescein-based profluorophores. | ||
Despite the extensive use of FDA and its derivatives in biological experiments, fluorescein diesters suffer from two juxtaposed problems. Fluorescein displays a relatively low phenolic pKa value of 6.4, causing instability of the ester bonds in aqueous solution.4 These low pKa values are, however, necessary for utility because the conjugate base of the fluorophore is much more fluorescent than the protonated form.11 Substitution on the fluorescein xanthene ring system can affect this pKa. For example, 2′,7′-difluorofluorescein (1b; Scheme 1) exhibits a phenolic pKa of 4.7 and is thus less sensitive to biologically-relevant pH fluctuations.12 This fluorine substitution, however, leads to even lower chemical stability for 2′,7′-difluorofluorescein diacetate (DFFDA; 2b) in aqueous solution (vide infra).
High chemical stability of fluorogenic probes is vital for certain experiments, as spontaneous hydrolysis compromises the ability to assess enzymatic activity, raises background fluorescence, and diminishes membrane permeance. One strategy to improve the stability of fluorogenic substrates involves insertion of a self-immolative chemical functionality between the fluorophore and the enzyme-reactive moiety. Enzymatic catalysis yields an intermediate that undergoes rapid decomposition to release the free fluorophore. This approach has been used to construct substrates for various hydrolytic enzymes.13–16Esterase substrates, in particular, benefit from this strategy, as it allows a labile ester to be replaced with a more stable functional group. For example, a 2-oxopropyloxy spacer endows umbelliferone-based fluorogenic lipase and esterase substrates with high stability.17 We have also employed this strategy, using the “trimethyl lock” motif to mask amine-containing fluorophores. These “profluorophores” possess high chemical stability, and are unmasked by esterases efficiently both in vitro and in cellulo.4,18–21
Many of the existing auto-immolative linkers add undesirable size and synthetic complexity to the probe or cannot offer significant advantages to phenolic fluorophores, such as fluorescein. Instead, we envisioned replacing the ester functionalities in a fluorescein diacetate with acetoxymethyl (AM) ethers. Such insertion of a simple oxygen–methylene group into the fluorophore–acetate bond should endow the compound with increased stability. Hydrolysis of the enzyme-labile moiety in these AM-containing substrates results in a hemiacetal that decomposes spontaneously to liberate the phenolic fluorophore. This modification alters significantly the leaving group pKa (formaldehyde hydrate has a pKa of 12.78, ref. 22), thereby increasing chemical stability. The modification should also insulate the labile ester group from the dye, allowing flexibility in dye type.
A well-known application of the acetoxymethyl strategy is the masking of carboxyl groups. AM esters were first used to increase the cell-permeability of certain antibiotics23,24 and this is now a well-known prodrug strategy.8 Tsien, Schultz, and their coworkers established AM esters as an effective and innocuous means to deliver carboxylate-containing ion chelators and even phosphorylated molecules into cells.5,23,24
AM ethers are less common. This motif has been used in prodrug strategies,25–28 to mask the fluorophoric portions of cell-permeable fluorescent ion indicators3,6 or labels,29 and to create fluorogenic enzyme substrates. Reymond and coworkers established that monoacyloxymethyl ethers of umbelliferone30 or fluorescein31,32 could be useful fluorogenic enzyme substrates with significantly improved chemical stability. Such compounds allow the accurate assessment of enzymatic activity in vitro. Unfortunately, these probes cannot be used to assess esterase activityin cellulo due to suboptimal wavelengths and relatively modest changes in fluorescence upon unmasking.
Here, we use the AM ether system to build fully masked, diacyloxymethyl ether substrates based on fluorescein and 2′,7′-difluorofluorescein (“Oregon Green”). We first determine experimental conditions that enable the efficient synthesis of such compounds from accessible chemical reagents in a single step. We then assess their hydrolytic stability, enzymatic turnover, and utility in live human cells. We then extend the acetoxymethyl ether strategy to the fluorophores “Tokyo Green”![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 33 and resorufin and compare these substrates to those based on the fluoresceins. Our results show the AM ether profluorophores—particularly those based on the classic fluoresceins—possess much greater chemical stability than simple fluorophore esters, but are still unmasked efficiently by esterases both in vitro and in cellulo.
33 and resorufin and compare these substrates to those based on the fluoresceins. Our results show the AM ether profluorophores—particularly those based on the classic fluoresceins—possess much greater chemical stability than simple fluorophore esters, but are still unmasked efficiently by esterases both in vitro and in cellulo.
Results and discussion
Chemical stability and reactivity of fluorescein diacetate derivatives
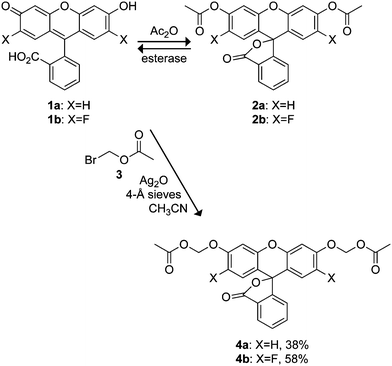
The chemical stability of both fluorescein diacetate (2a) and 2′,7′-difluorofluorescein diacetate (2b) was assessed by incubating these compounds in aqueous solution and monitoring the accretion of fluorescence over time. Both compounds exhibited low initial fluorescence signals in Dulbecco's phosphate-buffered saline (DPBS) but underwent slow, spontaneous hydrolysis (Fig. 1A). As expected from the leaving group pKa values, the hydrolysis of fluorinated acetate ester 2b is faster than that of 2a. The two compounds exhibit poor stability in Dulbecco's modified Eagle's medium containing fetal bovine serum (DMEM–FBS; Fig. 1B). Here, DFFDA (2b) exhibited an apparent34 half-life of 6 min and FDA (2a) was hydrolyzed completely in hours (t1/2 = 1.5 h).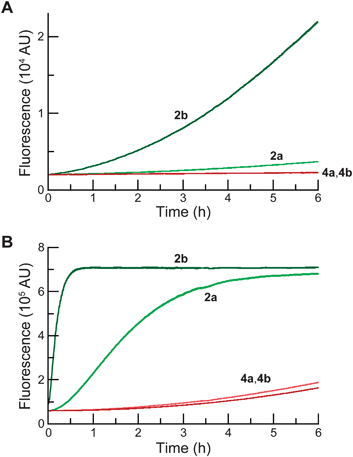 | ||
| Fig. 1 Time course for the spontaneous generation of fluorescence (λex 496 nm, λem 520 nm) from acetate esters 2a and 2b (25 nM), and AM ethers 4a and 4b (25 nM). (A) In DPBS. (B) In DMEM containing 10% v/v FBS. | ||
Although FBS likely contains low levels of serum esterases,5,6,35 we suspected the observed instability is due in part to the non-specific reactivity of the acyl groups with the ample nucleophiles in the tissue culture milieu. To test this hypothesis, we incubated acetate esters 2a and 2b with Ac-Arg-Phe-Met-Trp-Met-Lys-NH2. This hexapeptide contains a single Lys residue along with charged and chromophoric residues that allow facile separation and detection by tandem liquid chromatography–mass spectrometry (LC–MS). Analysis of this reaction mixture showed significant acylation of the peptide, with fluorinated 2b being more reactive than 2a (see: Fig. S1 in the ESI). These data reveal a critical limitation of acetate ester dyes in the accurate monitoring of esterase activity, providing an additional incentive to prepare chemically stable esterase substrates.
Synthesis of fluorescein AM ethers
The synthesis of diacetoxymethyl ethers of fluorescein derivatives was complicated by several factors. Previous syntheses of fluorogenic AM ethers involve reaction of the phenolic fluorophore with a halomethyl ester such as bromomethyl acetate using either NaH30–32 or N,N-diisopropylethylamine (DIEA).29Halomethyl esters possess two electrophilic centers: the halide-bearing carbon and the carbonyl carbon. Thus, base-mediated installation of a single AM ether leads to a mixture of ester and ether products and can result in low conversion to the desired compound.26,32 Based on the poor reported yield (∼10%) of monosubstituted acetoxymethyl fluorescein,32 we suspected the preparation of disubstituted AM ether profluorophores would require optimization of reaction conditions.An additional caveat involves the complexity of the fluorescein molecule. In solution, xanthene dyes such as fluorescein exist as an equilibrium mixture of quinoid and lactone forms, their distribution depending on the environment.21,36,37 Treatment of fluorescein with an alkylating agent typically gives an ether–ester as the major product.38–40 Thus, despite the apparent simplicity of the desired molecules, the bifurcated reactivity of the halomethyl acetate in tandem with the fluorescein equilibrium issue leads to several reaction products.
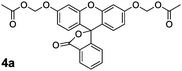
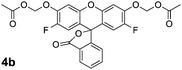
Based on the prodrug25–27 and profluorophore5,41–45 literature, we screened combinations of solvent and base to find optimal conditions for the synthesis of the desired AM ether products 4a and 4b from dyes 1a and 1b with commercially available bromomethyl acetate (3). As shown in Scheme 1, we found the use of Ag2O in acetonitrile with molecular sieves to be effective for the synthesis of both fluorescein derivatives 4a and 4b in moderate isolated yield. Use of a heterogeneous Ag(I) salt should favour the desired alkylation reaction over acylation.46 The polar aprotic acetonitrile appears to maintain the fluoresceins in their closed lactone form. The superior yield obtained for compound 4b relative to 4a can be explained, in part, by the lower nucleophilicity of the relevant phenolic oxygen leading to a tighter SN2 transition state and subsequent increase in alkylation over acylation.47
We note that DMF/DIEA gave only trace amounts (>3%) of 4a and a 19% yield of 4b. DMF–toluene mixtures with Ag2O gave low yields of 5 and 15% for 4a and 4b, respectively. We did, however, observe success in the synthesis of the fluorescein diacetoxymethyl ether (4a) by using a phase-transfer reaction protocol,27 which gave the desired molecule in 31% isolated yield. The phase-transfer recipe gave only a poor yield (6%) of 4b. We conclude that our reaction conditions constitute a marked improvement in the route to fluorescein AM ethers compared to typical methods.30,32
Chemical stability and reactivity of AM ethers
As with fluorescein diacetate (2a) and 2′,7′-difluorofluorescein diacetate (2b), the chemical stability of both fluorescein diacetoxymethyl ether (4a) and 2′,7′-difluorofluorescein diacetoxymethyl ether (4b) profluorophores was assessed in aqueous solution. These two compounds showed low initial fluorescence signals in Dulbecco's phosphate-buffered saline (DPBS; Fig. 1A), indicating that both acylation and alkylation of the phenolic groups in fluorescein lock the molecule into the colorless, nonfluorescent lactone form. The AM ether profluorophores (4a and 4b) suffered hydrolysis slower than did their acetate ester congeners (2a and 2b) in either DPBS (Fig. 1A) or DMEM–FBS (Fig. 1B). Longer incubation in a microplate format revealed the apparent34 half-life for both 4a and 4b to be 32 h in DMEM–FBS (see: Fig. S2 in the ESI).We also incubated these profluorophores with the lysine-containing hexapeptide. In contrast to the acetate esters 2a and 2b, we observed no measurable acylation by 4a and 4b of the hexapeptide (see: Fig. S1 in the ESI). The similarity in the chemical stability of the AM ether dyes supports our hypothesis that the acetoxymethyl group insulates the ester bond, and thereby abolishes the effect of the fluoro groups on the chemical stability of the substrate. Profluorophore 4b and its derivatives could prove to be especially valuable to chemical biologists as a fluorogenic esterase substrate exhibiting higher chemical stability than fluorescein diacetate (2a), but releasing difluorofluorescein, a pH-insensitive dye with high photostability.12
Enzyme kinetics
Having established the enhanced chemical stability of profluorophores 4a and 4b, we sought to assess the enzymatic reactivity of these potential enzyme substrates in vitro, comparing them to the parent diacetate dyes. Incubation of each of these four compounds with porcine liver esterase (PLE) elicited a rapid increase in fluorescence intensity. Fluorescein diacetate (2a) proved to be the best substrate, with kcat/KM = 1.4 × 106M−1s−1 and KM = 11 μM (see: Fig. S3 in the ESI). These data are in gratifying agreement with values reported previously.48 Surprisingly, difluorofluorescein diacetate (2b) exhibited slower turnover, giving apparent34 kinetic constants of kcat/KM = 2.9 × 105M−1s−1 and KM = 5.3 μM. The two AM ether substrates behaved similarly, with kcat/KM = 6.8 × 105M−1s−1 and KM = 3.2 μM for 4a, and kcat/KM = 3.8 × 105M−1s−1 and KM = 4.9 μM for 4b. We conclude that the AM ether modification does not greatly diminish the performance of these substrates in vitro.Cellular imaging
Then, we assessed the utility of the AM ether-masked dyes in human cells. To determine whether these compounds could be unmasked by endogenous esterase activity, we obtained widefield (left panel) and confocal (right panel) microscopy images of live, unwashed HeLa cells incubated with fluorescein diacetate (2a; Fig. 2A), fluorescein diacetoxymethyl ether (4a; Fig. 2B), 2′,7′-difluorofluorescein diacetate (2b; Fig. 2C), or 2′,7′-difluorofluorescein diacetoxymethyl ether (4b; Fig. 2D). All four experiments produced bright cellular staining after 20 min, suggesting that each molecule is internalized and unmasked by cellular esterases. The widefield microscopy images show the high relative extracellular background for the diacetate dyes 2a and 2b, in accordance with the chemical stability data (Fig. 1). AM ethers 4a and 4b exhibited significantly lower extracellular fluorescence. The confocal images of unwashed cells show that the AM ether substituent does not affect subcellular partitioning. Thus, probes 4a and 4b could be useful substitutes for diacetate dyes in biological experiments where extracellular washing is either undesirable (e.g., high-throughput screening) or impossible (e.g., tissue and in vivo imaging). These stable substrates should also provide a more accurate assessment of cellular esterase activity. | ||
| Fig. 2 Live, unwashed HeLa cells incubated with acetate esters 2a and 2b, AM ethers 4a and 4b (10 μM) in DMEM–FBS for 20 min and counterstained with Hoechst 33342 (5% v/v CO2(g), 100% humidity); left panel: wide-field microscopy, right panel: confocal microscopy. (A) Acetate ester 2a (FDA). (B) AM ether 4a. (C) Acetate ester 2b (DFFDA). (D) AM ether 4b. Scale bars: 10 μm. | ||
Tokyo Green substrates
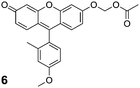
Fluorescein substrates 2a–b and 4a–b each contain two substrate moieties per molecule. To investigate the effect of monosubstitution versus disubstitution on stability and enzyme kinetics, we prepared profluorophores based on Tokyo Green (5)33 as shown in Scheme 2. Reaction of 5 with bromomethyl acetate (3) in the presence of Hünig's base gave the desired AM ether 6 in good yield (Scheme 2). The Tokyo Green acetate 7 was also prepared in 84% yield via microwave-assisted synthesis in neat acetic anhydride. Unlike fluorescein, the Tokyo Green molecule does not bear an ortho carboxyl group on its pendant phenyl ring, thus circumventing the complex lactone–quinoid equilibria. Instead, this electron-rich aromatic substituent facilitates photoinduced electron transfer (PeT) that quenches fluorescence when the xanthenyl moiety is protonated or alkylated.33,49 We reasoned that the acetoxymethyl ether strategy could suppress the fluorescence of this dye, providing a stable substrate that would be unmasked in a single step. | ||
| Scheme 2 Tokyo Green-based profluorophores. | ||
Evaluation of the stability of the Tokyo Green-based profluorophores was performed as with the other fluorescein-based profluorophores. As expected, AM ether 6 proved to be more resistant to hydrolysis than acetate ester 7 in both DPBS (Fig. 3A) and DMEM–FBS (Fig. 3B). Tokyo Green derivatives underwent spontaneous hydrolysis faster than did the classic fluorescein derivatives. Data from a longer incubation revealed AM ether 6 to have a half-life of 5 h in DMEM–FBS (see: Fig. S4 in the ESI). Acetate ester 7 showed little chemical stability in this solution, having t1/2 < 2 min. Acetate ester 7 also acylated the hexapeptide readily, whereas AM ether 6 did not exhibit any appreciable reaction (see: Fig. S5 in the ESI).
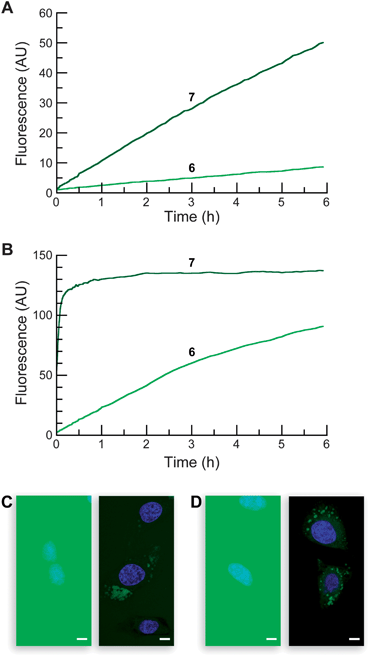 | ||
| Fig. 3 Properties of Tokyo Green-based profluorophores. Time course for the spontaneous generation of fluorescence (λex 496 nm, λem 520 nm) from acetate ester 7 and AM ether 6 (25 nM). (A) In DPBS. (B) In DMEM containing 10% v/v FBS. (C–D) Live-cell imaging experiments with substrates 7 and 6 (10 μM) incubated for 20 min in DMEM–FBS and counterstained with Hoechst 33342 (5% v/v CO2(g), 100% humidity); left panel: wide-field microscopy, right panel: confocal microscopy. (C) Acetate 7. (D) AM ether 6. Scale bar: 10 μm. | ||
The Tokyo Green derivatives proved to be excellent substrates for PLE, giving kinetic constants of kcat/KM = 3.2 × 106M−1s−1 and KM = 12 μM for acetate ester 7, and kcat/KM = 4.2 × 106M−1s−1 and KM = 13 μM for AM ether 6 (see: Fig. S3 in the ESI). Incubation of profluorophores 6 and 7 with living cells shows extensive intra- and extracellular fluorescence in the widefield images (Fig. 3C–D, left panel). Confocal imaging shows moderate cellular staining only with the AM ether 6, in line with the stability data (Fig. 3C–D, right panel). The widespread signal is likely due to poor cellular retention of the unmasked Tokyo Green 5; cellular fluorescence diminishes quickly with washing (data not shown).
These data imply that, for many experiments, the disubstituted dyes 2a–b and 4a–b could prove superior to the monosubstituted congeners 7 and 6. The monosubstituted dyes appear less stable in both DPBS and DMEM–FBS. The kcat/KM values of the monosubstituted dyes are not substantially higher than those of the disubstituted substrates. In addition, the classic fluorescein system contains a shrouded carboxyl group that is unmasked concomitantly with the ester groups, leading to improved cellular retention relative to the Tokyo Green substrates. Still, substrate 6 could find use in experiments where rapid assessment of esterase activity is required without the need for cellular imaging.
Resorufin acetoxymethyl ethers
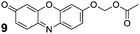
Resorufin (8) exhibits excitation and emission wavelengths that are red-shifted relative to fluorescein (λex = 572 nm, λem = 585 nm).50 Substitution on the phenolic group of resorufin elicits a hypsochromic shift and a dramatic decrease in quantum yield. Alkylated and glycosylated variants of resorufin are used extensively as enzyme substrates.51,52 Based on this precedence, resorufin seemed to be a good candidate for the acetoxymethyl ether strategy. We prepared profluorophore 9 through both Ag2O-mediated alkylation and phase-transfer conditions. We note the poor solubility of resorufin in organic solvents—a pernicious problem with this fluorophore—resulted in a low yield of 33% for the Ag(I)-mediated reaction. Phase-transfer conditions gave a markedly better yield of 73% (Scheme 3).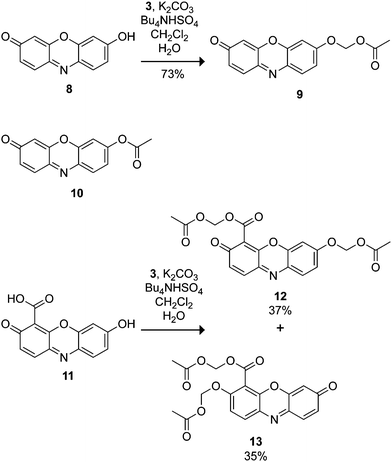 | ||
| Scheme 3 Resorufin-based profluorophores. | ||
We examined the stability of AM ether 9 and commercial resorufin acetate (10). Profluorophore 10 has long been recognized as an effective esterase substrate,53 but the utility of this compound is limited by its poor stability in aqueous solution.54 The chemical stability of resorufin derivatives 9 and 10 was similar to that of the Tokyo Green-based substrates, as expected from their analogous structures. As with the other substrates, AM ether 9 exhibited higher stability than did acetate 10 in DPBS (Fig. 4A). In DMEM–FBS, the observed half-lives were 3 min and 2 h for substrates 10 and 9, respectively (Fig. 4B). Following the trend of the aforementioned substrates, compound 10 served as an acyl donor for the lysine-containing peptide, whereas AM ether dye 9 did not modify the peptide (see: Fig. S6 in the ESI). Compounds 9 and 10 were substrates for PLEin vitro. The kinetic constants were kcat/KM = 3.1 × 106M−1s−1 and KM = 16 μM for profluorophore 10, and kcat/KM = 3.6 × 106M−1s−1 and KM = 21 μM for profluorophore 9 (see: Fig. S3 in the ESI). These kinetic constants are similar to those of the other monosubstituted profluorophores, 6 and 7.55
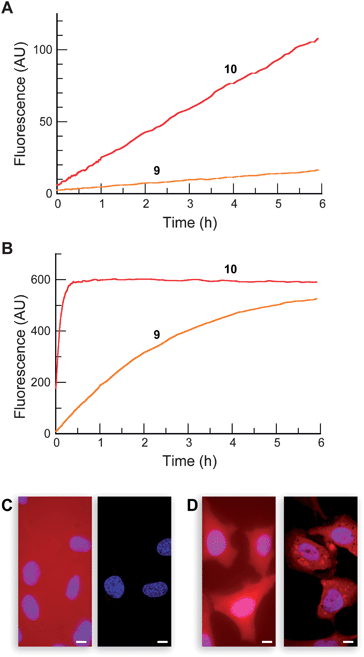 | ||
| Fig. 4 Properties of resorufin-based profluorophores. Time course for the spontaneous generation of fluorescence (λex 572 nm, λem 585 nm) for acetate ester 10 and AM ether 9 (25 nM) in (A) DPBS and (B) DMEM containing 10% v/v FBS. (C–D) Live-cell imaging experiments with substrates 9 and 12 (10 μM) incubated for 20 min in DMEM–FBS and counterstained with Hoechst 33342 (5% v/v CO2(g), 100% humidity); left panel: wide-field microscopy, right panel: confocal microscopy. (C) AM ether 9. (D) AM ether/AM ester 12. Scale bar: 10 μm. | ||
We exposed live human cells to profluorophores 9 and 10. The diminished cellular autofluorescence at this longer excitation wavelength allowed lower substrate concentrations (2 μM) to be used relative to the green profluorophores. Acetate 10 stained the field nondiscriminately with no cellular staining (data not shown). As with compound 6, fluorescence microscopy imaging experiments with AM ether 9 showed substantial extracellular background in the widefield images and low staining in the confocal images (Fig. 4C). These data suggest that one anionic charge is insufficient to retain the fluorescent molecule inside cells.
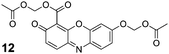
We therefore subjected 4-carboxyresorufin (11)56 to the AM ether strategy, using phase-transfer conditions to prepare the isomeric substrates 12 and 13 in 37 and 35% yield, respectively. We used compound 12 in subsequent experiments. As with AM ether 9, incubation with peptide confirmed that substrate 12 does not serve as a lysine acylating agent (see: Fig. S6 in the ESI). We then incubated this compound with live cells, reasoning the extra carboxyl group would alter the partitioning of the dyein cellulo. In contrast to substrate 9, incubation with the same concentration of the 4-carboxyresorufin derivative 12 resulted in bright red cellular staining with relatively low background fluorescence (Fig. 4D), showcasing the effect of a single carboxylate moiety on the cellular retention of this dye. This compound could find use in multicolour cell biological experiments, as fluorogenic esterase substrates with excitation wavelengths >500 nm remain notably sparse.2,19
Conclusions
The insertion of a simple oxygen–methylene group into a fluorophore ester bond endows molecules with high chemical stability. Importantly, we demonstrate this modification insulates the ester moiety from the fluorophore structure, allowing the stable masking of disparate dyes to create a portfolio of substrates with different chemical and spectroscopic properties. We have discovered reaction conditions, such as the use of Ag(I) salts in acetonitrile or phase-transfer conditions, that enable the synthesis of problematic compounds such as fluorescein and resorufin derivatives in moderate yields. Each of the described novel AM ether profluorophores (4a, 4b, 6, 9, and 12; Table 1) should be useful for assessing esterase activity in different biochemical or biological contexts. The stability and nascent polarity of substrates 4a, 4b, and 12 make these compounds useful for examining hydrolytic enzymes in cellular fluorescence imaging experiments.| Profluorophore | k cat/KMa/M−1s−1 | λ ex b/nm | λ em b/nm | HeLa-cell retention? |
|---|---|---|---|---|
| a Values of kcat/KM are for catalytic hydrolysis by pig liver esterase, and are apparent values for compounds 4a and 4b.34 ND: not determined. b Values of λex and λem are for unmasked profluorophores. | ||||
|
6.8 × 105 | 496 | 520 | Yes |
|
3.8 × 105 | 496 | 520 | Yes |
|
4.2 × 106 | 496 | 520 | No |
|
3.6 × 106 | 572 | 585 | No |
|
ND | 572 | 585 | Yes |
Numerous applications of these stable substrates are possible. Substitution of the acetyl functionality with other acyl groups32 could allow the assessment of esterase activityin cellulo. Such experiments could facilitate the discovery of new ester motifs useful for the efficient delivery of small molecules to cells. In addition, the distribution of esterases between the secretory pathway and cytosol is unknown,8 and this platform could be used to map esterase activity within cells as well as tissues.57 Bioconjugation with derivatives of these compounds could facilitate biomolecular imaging experiments.4 Use of phosphoryl moieties in place of acyl groups58 could provide stable substrates for phosphatases. Overall, this strategy could supplement or supplant fluorophore acetate esters, allowing facile means for illuminating biochemical and biological systems.
Experimental
Chemical synthesis: General
2′,7′-Difluorofluorescein (1b; ref. 12), Tokyo Green (7; ref. 33), and 4-carboxyresorufin (11; ref. 56) were synthesized as described previously. Preactivated, powdered 4-Å molecular sieves from Aldrich were used as received. Anhydrous solvents were drawn from a Baker CYCLE-TAINER solvent delivery system or Aldrich Sure-Seal bottles. All other reagents were obtained from Sigma-Aldrich or Fisher Scientific, and used without further purification.Microwave reactions were performed in an Explorer 48 system from CEM. Thin-layer chromatography was performed by using aluminium-backed plates coated with silica gel containing F254 phosphor and visualized by UV-illumination or staining with I2, ceric ammonium molybdate, or phosphomolybdic acid. Flash chromatography was performed by using open columns loaded with silica gel-60 (230–400 mesh) on a FlashMaster Solo system (Argonaut) with Isolute Flash Si II columns (International Sorbent Technology), or on an Isolera 4 system with SNAP columns (Biotage). The term “high vacuum” refers to a vacuum (≤1 mm Hg) achieved by a mechanical belt-drive oil pump. The term “concentrated under reduced pressure” refers to the removal of solvents and other volatile materials by using a rotary evaporator at variable pressure (controlled diaphragm pump or water aspirator; >1 mm Hg) while maintaining the water-bath temperature below 40 °C. The term “concentrated under high vacuum” refers to the removal of solvents and other volatile materials by using a rotary evaporator at high vacuum while maintaining the water-bath temperature below 40 °C.
NMR spectra were obtained with a Bruker 400 MHz Avance spectrometer at the NMR Facility at Madison (NMRFAM) or on a Bruker 400 MHz Avance-II+spectrometer at the Janelia Farm Research Campus. 1H and 13C NMR spectra were referenced to TMS or residual solvent peaks. 19F NMR spectra were referenced to CFCl3. Mass spectrometry was performed with a Micromass LCT (electrospray ionization, ESI) mass spectrometer in the Mass Spectrometry Facility in the Department of Chemistry.
Fluorescein diacetoxymethyl ether (4a)
2′,7′-Difluorofluorescein diacetoxymethyl ether (4b)
2′,7′-Difluorofluorescein (1b, 133 mg, 0.361 mmol), powdered 4-Å molecular sieves (300 mg), and anhydrous Ag2O (209 mg, 0.903 mmol) were suspended in anhydrous CH3CN (6 mL) under Ar(g). Bromomethyl acetate (3, 0.142 mL, 1.44 mmol) was added dropwise, and the reaction mixture was stirred for 48 h. The reaction mixture was then diluted with CH2Cl2 and filtered through a pad of celite. The solution was concentrated under reduced pressure to give an orange oil. Purificationviacolumn chromatography (silica gel, 0 → 40% v/v EtOAc in hexanes containing constant 40% v/v CH2Cl2 as cosolvent) afforded profluorophore 4b as a white crystalline solid (107 mg, 58%). 1H NMR (400 MHz, CDCl3) δ (ppm): 8.07 (d, J = 7.6 Hz, 1H), 7.5 (ddd, J = 7.6, 7.4, 1.2 Hz, 1H), 7.70 (ddd, J = 7.5, 7.3, 1.0 Hz, 1H), 7.19 (d, J = 7.5 Hz, 1H), 7.10 (J = 7.0 Hz, 2H), 6.52 (d, J = 10.7 Hz, 2H), 5.83 (ABq, J = 6.4 Hz, 4H), 2.19 (s, 6H). 13C NMR (100 MHz, CDCl3) δ (ppm): 169.50, 168.54, 151.86, 150.53, 148.08, 147.48, 146.65, 146.55, 135.51, 130.45, 126.21, 125.54, 123.75, 114.73, 114.53, 112.42, 112.36, 105.54, 85.61, 81.62, 20.85. 19F NMR (400 MHz, CDCl3) δ (ppm): −136.92 (dd, J = 10.9, 6.8 Hz). ESIMS [M + Na]+ calculated, 535.0812; found, 535.0820.Tokyo Green acetoxymethyl ether (6)
Tokyo Green (5, 50 mg, 0.150 mmol), bromomethyl acetate (3, 74 μL, 0.75 mmol), and Hünig's base (158 μL, 0.90 mmol) were dissolved in anhydrous DMF (2.0 mL) under N2(g), and the reaction mixture was stirred for 24 h. The reaction mixture was then concentrated under high vacuum to give an orange solid. Purificationviacolumn chromatography (silica gel, first column: 50% v/v EtOAc in CH2Cl2; second column: 2 → 4% v/v MeOH in CH2Cl2) afforded compound 6 as an orange solid (48 mg, 79%). 1H NMR (400 MHz, CDCl3) δ (ppm): 7.11 (d, J = 2.4 Hz, 1H), 7.06 (d, J = 8.2 Hz, 1H), 7.05 (d, J = 9.1 Hz, 1H), 6.99 (d, J = 9.9 Hz, 1H), 6.93–6.87 (m, 2H), 6.85 (dd, J = 9.1, 2.4 Hz, 1H), 6.55 (dd, J = 9.7, 1.9 Hz, 1H), 6.42 (d, J = 1.9 Hz, 1H), 5.83 (s, 2H), 3.88 (s, 2H), 2.14 (s, 2H), 2.03 (s, 2H). 13C NMR (100 MHz, CDCl3) δ (ppm): 185.84, 169.52, 160.73, 160.38, 158.77, 154.05, 148.95, 137.81, 130.69, 130.34, 130.29, 129.74, 124.31, 119.44, 116.12, 116.01, 113.78, 111.56, 105.85, 102.71, 84.38, 55.33, 20.79, 19.95. ESIMS [M + H]+ calculated, 405.1333; found, 405.1325.Tokyo Green acetate (7)
Tokyo Green (5, 20 mg, 0.060 mmol) was dissolved in acetic anhydride (2.0 mL) in a 10-mL microwave reaction vial. This vial was heated in a microwave oven to 90 °C for 10 min (200 W with cooling). The reaction mixture was concentrated under high vacuum to give an orange solid. Purificationviacolumn chromatography (silica gel, 25 → 75% v/v EtOAc in hexanes containing constant 20% v/v CH2Cl2 as cosolvent) afforded profluorophore 7 as an orange solid (19 mg, 84%). 1H NMR (400 MHz, CDCl3) δ (ppm): 7.27 (d, J = 2.1 Hz, 1H), 7.12 (d, J = 8.8 Hz, 1H), 7.09 (d, J = 8.8 Hz, 1H), 7.02 (d, J = 9.8 Hz, 1H), 6.93 (m, 2H), 6.58 (dd, J = 9.8, 2.0 Hz, 1H), 6.43 (d, J = 2.1 Hz, 1H), 3.90 (s, 3H), 2.36 (s, 3H), 2.07 (s, 3H). 13C NMR (100 MHz, CDCl3) δ (ppm): 186.04, 168.58, 160.52, 158.59, 154.16, 153.13, 148.23, 137.97, 130.90, 130.75, 130.47, 129.22, 124.21, 120.87, 118.86, 118.30, 116.13, 111.70, 110.24, 106.16, 55.39, 21.17, 20.05. ESIMS [M + H]+ calculated, 375.1227; found, 375.1228.Resorufin acetoxymethyl ether (9)
:3:2 → 3:4:3 v/v/v hexanes:EtOAc:CH2Cl2; second column: 10% v/v CH3CN in CHCl3) to give compound 9 as a red-orange solid (87 mg, 33%). Analytical data matched the material prepared with Method 1.
4-Carboxyresorufin acetoxymethyl ether, acetoxymethyl ester (12 and 13)
4-Carboxyresorufin (11, 77 mg, 0.299 mmol) and anhydrous K2CO3 (248 mg, 1.80 mmol) were dissolved in water (2.5 mL) to give a dark red solution. Tetrabutylammonium bisulfate (203 mg, 0.599 mmol) in CH2Cl2 (1.0 mL) was added, followed by bromomethyl acetate (3, 235 μL, 2.40 mmol) in CH2Cl2 (1.5 mL). The reaction mixture was stirred at high speed (∼600 rpm) at ambient temperature for 24 h. The reaction mixture was then diluted with H2O and CH2Cl2, and the layers were separated. The aqueous layer was extracted with CH2Cl2, and the combined organics were washed with H2O and saturated brine. The organic layer was dried over anhydrous MgSO4(s) and concentrated under reduced pressure to give an orange oil. Purificationviacolumn chromatography (silica gel, 25 → 50% EtOAc in hexanes containing constant 20% v/v CH2Cl2) gave compound 12 as a red-orange solid (45 mg, 37%) and compound 13 as a brown crystalline solid (32 mg, 35%). Compound 12: 1H NMR (400 MHz, CDCl3) δ (ppm): 7.83 (d, J = 8.7 Hz, 1H), 7.50 (d, J = 10 Hz, 1H), 7.17–7.10 (m, 2H), 6.90 (d, J = 10 Hz, 1H), 6.03 (s, 2H), 5.87 (s, 2H), 2.22 (s, 3H), 2.19 (s, 3H). 13C NMR (100 MHz, CDCl3) δ (ppm): 181.40, 169.60, 169.39, 161.91, 160.80, 147.73, 144.87, 144.68, 134.71, 134.28, 131.99, 129.34, 115.75, 110.90, 102.60, 84.54, 79.91, 20.75 (2C). ESIMS [M + Na]+ calculated, 424.0640; found, 424.0631. Compound 13: 1H NMR (400 MHz, CDCl3) δ (ppm): 7.84 (d, J = 9.0 Hz, 1H), 7.40 (d, J = 10 Hz, 1H), 7.16 (d, J = 9.0 Hz, 1H), 6.84 (dd, J = 9.9, 2.0 Hz, 1H), 6.30 (d, J = 2.0 Hz, 1H), 6.00 (s, 2H), 5.84 (s, 2H), 2.21 (s, 3H), 2.15 (s, 3H). 13C NMR (100 MHz, CDCl3) δ (ppm): 186.14, 169.53, 169.25, 161.44, 157.09, 148.63, 147.61, 142.32, 135.06, 134.75, 133.17, 129.06, 111.75, 111.61, 107.89, 84.96, 79.86, 20.77, 20.69. ESIMS [M + Na]+ calculated, 424.0640; found, 424.0641. Identity of isomers was confirmed by comparing the 2D NOESY spectra of compounds 9, 12, and 13 (see: ESI).Biochemistry: General
Dulbecco's phosphate-buffered saline (DPBS), Dulbecco's modified Eagle's medium (DMEM) and fetal bovine serum (FBS) were from Invitrogen. 2′,7′-Difluorofluorescein diacetate (2b) was synthesized as described previously.12 All other reagents were from Sigma-Aldrich or Fisher Scientific. HEPES buffer at pH 7.3 was prepared from a 1 M stock solution from Fisher.Fluorometry
Fluorometric measurements were made with a QuantaMaster1 photon-counting spectrofluorometer from Photon Technology International or an Eclipse spectrofluorometer from Varian, both equipped with sample stirring. Sample solutions were in fluorescence-grade quartz or glass cuvettes from Starna Cells, or fluorescence-grade polystyrene cuvettes from Perfector Scientific. All measurements were recorded at ambient temperature (23 ± 2 °C), and buffers were not degassed prior to measurements.Chemical stability and reactivity
Long-term stability studies (>12 h) were performed in black, clear-bottom, 96-well polystyrene microplates from Corning (product number 3651) sealed with SealPlate Film from PGC Scientifics. Plates were read from the bottom on a FlexStation 3 from Molecular Devices. Values of t½ (= ln2/k) were determined by fitting the data to a single exponential decay with eqn (1):| F = (F0 − Fmax)e−kt + Fmax | (1) |
Non-specific chemical reactivity was assessed by incubating profluorophore (100 μM) with Ac-Arg-Phe-Met-Trp-Met-Lys-NH2 (1.0 mg mL−1; Bachem; product number H-1994) in 10 mM HEPES buffer, pH 7.3, for 2 h at ambient temperature. The reaction mixture was analyzed on an Agilent 1200 HPLC system equipped with a G1315B diode array detector and a 6130 mass spectrometry detector using a 4.6 × 150 mm Eclipse XDB-C18 column with a gradient of 10 → 95% v/v CH3CN in H2O containing 0.1% v/v formic acid over 20 min.
Enzyme kinetics
Compounds were prepared as stock solutions in anhydrous DMSO and diluted such that the DMSO concentration did not exceed 1.5% v/v. Porcine liver esterase (PLE, MW = 163 kDa, ref. 59) was obtained from Sigma-Aldrich (product number E2884; lot 129K7010) as a suspension in 3.2 M (NH4)2SO4, and was diluted to appropriate concentrations in HEPES buffer, pH 7.4, before use in Protein LoBind tubes from Eppendorf. Kinetic parameters were calculated by fitting the data to the Michaelis–Menten equation with GraphPad Prism software.Cell preparation and imaging
HeLa cells were plated in a Nunc Lab-Tek II 8-well Chamber Coverglass (Fisher Scientific) and grown to 70–80% confluence at 37 °C in DMEM containing FBS (10% v/v). For microscopy, the medium was exchanged with DMEM–FBS containing substrates 2a, 2b, 4a, 4b, 6, 9, or 12 (2 or 10 μM), and cells were incubated for 20 min at 37 °C. Nuclei were stained by the addition of Hoechst 33342 (2 μg mL−1) during the final 5 min of incubation. Cells were imaged on an Olympus IX81 widefield microscope or a Zeiss 510-Meta confocal microscope. Cellular intensity was normalized for each set of fluorophores by using neutral density filters (widefield) or by adjusting laser power (confocal).Acknowledgements
We are grateful to Z. J. Diwu, J. B. Binder, R. J. Johnson, and K. J. Kolonko for contributive discussions, and A. C. Arnold for assistance with fluorescence microscopy. L.D.L. was supported by Biotechnology Training Grant T32 GM08349 (NIH) and an ACS Division of Organic Chemistry Fellowship sponsored by Genentech. T.-Y.C. was supported by the Dr James Chieh-Hsia Mao Wisconsin Distinguished Graduate Fellowship. This work was supported by grant R01 CA073808 (NIH) and the Howard Hughes Medical Institute. NMRFAM was supported by grant P41 RR02301 (NIH).Notes and references
- L. D. Lavis and R. T. Raines, ACS Chem. Biol., 2008, 3, 142–155 CrossRef CAS.
- R. P. Haugland, M. T. Z. Spence, I. D. Johnson and A. Basey, The Handbook: A Guide to Fluorescent Probes and Labeling Technologies, 10th edn, Molecular Probes, Eugene, OR, 2005 Search PubMed.
- J.-P. Goddard and J.-L. Reymond, Curr. Opin. Biotechnol., 2004, 15, 314 CrossRef CAS.
- L. D. Lavis, T.-Y. Chao and R. T. Raines, ACS Chem. Biol., 2006, 1, 252–260 CrossRef CAS.
- R. Y. Tsien, Nature, 1981, 290, 527–528 CrossRef CAS.
- J. Kao, A. Harootunian and R. Tsien, J. Biol. Chem., 1989, 264, 8179–8184 CAS.
- B. Rotman and B. W. Papermaster, Proc. Natl. Acad. Sci. U. S. A., 1966, 55, 134–141 CrossRef CAS.
- B. Testa and J. M. Mayer, Hydrolysis in Drug and Prodrug Metabolism: Chemistry, Biochemistry, and Enzymology, Verlag Helvetica Chimica Acta, Zürich, Switzerland, 2003 Search PubMed.
- L. D. Lavis, ACS Chem. Biol., 2008, 3, 203–206 CrossRef CAS.
- A. Baeyer, Ber. Dtsch. Chem. Ges., 1871, 4, 555–558 CrossRef.
- L. D. Lavis, T. J. Rutkoski and R. T. Raines, Anal. Chem., 2007, 79, 6775–6782 CrossRef CAS.
- W.-C. Sun, K. R. Gee, D. H. Klaubert and R. P. Haugland, J. Org. Chem., 1997, 62, 6469–6475 CrossRef CAS.
- N. H. Ho, R. Weissleder and C. H. Tung, ChemBioChem, 2007, 8, 560–566 CrossRef CAS.
- Y. Meyer, J. Richard, M. Massonneau, P. Renard and A. Romieu, Org. Lett., 2008, 10, 1517–1520 CrossRef CAS.
- J. A. Richard, Y. Meyer, V. Jolivel, M. Massonneau, R. Dumeunier, D. Vaudry, H. Vaudry, P. Y. Renard and A. Romieu, Bioconjugate Chem., 2008, 19, 1707–1718 CrossRef CAS.
- X. Zhang, M. Waibel and J. Hasserodt, Chem.–Eur. J., 2010, 16, 792–795 CAS.
- R. Sicart, M. P. Collin and J.-L. Reymond, Biotechnol. J., 2007, 2, 221–231 Search PubMed.
- S. S. Chandran, K. A. Dickson and R. T. Raines, J. Am. Chem. Soc., 2005, 127, 1652–1653 CrossRef CAS.
- L. D. Lavis, T. Y. Chao and R. T. Raines, ChemBioChem, 2006, 7, 1151–1154 CrossRef CAS.
- M. M. Yatzeck, L. D. Lavis, T. Y. Chao, S. S. Chandran and R. T. Raines, Bioorg. Med. Chem. Lett., 2008, 18, 5864–5866 CrossRef CAS.
- R. W. Watkins, L. D. Lavis, V. M. Kung, G. V. Los and R. T. Raines, Org. Biomol. Chem., 2009, 7, 3969–3975 RSC.
- P. D. Taylor, Talanta, 1995, 42, 243–248 CrossRef CAS.
- C. Schultz, M. Vajanaphanich, A. T. Harootunian, P. J. Sammak, K. E. Barrett and R. Y. Tsien, J. Biol. Chem., 1993, 268, 6316–6322 CAS.
- C. Schultz, Bioorg. Med. Chem., 2003, 11, 885–898 CrossRef CAS.
- N. Bodor, K. B. Sloan, J. J. Kaminski, C. Shih and S. Pogany, J. Org. Chem., 1983, 48, 5280–5284 CrossRef CAS.
- H. Ouyang, R. T. Borchardt and T. J. Siahaan, Tetrahedron Lett., 2002, 43, 577–579 CrossRef CAS.
- J. D. Thomas and K. B. Sloan, Tetrahedron Lett., 2007, 48, 109–112 CrossRef CAS.
- J. Thomas and K. Sloan, Int. J. Pharm., 2008, 346, 80–88 CrossRef CAS.
- C. Uttamapinant, K. A. White, H. Baruah, S. Thompson, M. Fernández-Suárez, S. Puthenveetil and A. Y. Ting, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 10914–10919 CrossRef CAS.
- E. Leroy, N. Bensel and J.-L. Reymond, Bioorg. Med. Chem. Lett., 2003, 13, 2105–2108 CrossRef CAS.
- N. Bensel, M. T. Reymond and J.-L. Reymond, Chem.–Eur. J., 2001, 7, 4604–4612 CrossRef CAS.
- Y. Z. Yang, P. Babiak and J.-L. Reymond, Helv. Chim. Acta, 2006, 89, 404–415 CrossRef.
- Y. Urano, M. Kamiya, K. Kanda, T. Ueno, K. Hirose and T. Nagano, J. Am. Chem. Soc., 2005, 127, 4888–4894 CrossRef CAS.
- We use the term “apparent” because full fluorescence manifestation requires the removal of two AM ether moieties.
- B. P. Doctor, T. C. Chapman, C. E. Christner, C. D. Deal, D. M. De La Hoz, M. K. Gentry, R. A. Ogert, R. S. Rush, K. K. Smyth and A. D. Wolfe, FEBS Lett., 1990, 266, 123–127 CrossRef CAS.
- V. Zanker and W. Peter, Ber. Dtsch. Chem. Ges., 1958, 91, 572–580 CAS.
- I. S. Ioffe and V. F. Otten, Zh. Obshch. Khim., 1965, 1, 343–346 CAS.
- A. G. Miller, Anal. Biochem., 1983, 133, 46–57 CAS.
- B. A. Sparano, S. P. Shahi and K. Koide, Org. Lett., 2004, 6, 1947–1949 CrossRef CAS.
- L. Mugherli, O. Burchak, F. Chatelain and M. Balakirev, Bioorg. Med. Chem. Lett., 2006, 16, 4488–4491 CrossRef CAS.
- G. A. Krafft, W. R. Sutton and R. T. Cummings, J. Am. Chem. Soc., 1988, 110, 301–303 CrossRef CAS.
- T. J. Mitchison, J. Cell Biol., 1989, 109, 637–652 CrossRef CAS.
- R. P. Haugland, J. J. Naleway and Y.-Z. Zhang, U.S. Patent 5,208,148.
- K. Kasai, R. Uchida and N. Yamaji, Chem. Pharm. Bull., 1993, 41, 314–318 CAS.
- T. J. Mitchison, K. E. Sawin, J. A. Theriot, K. Gee, A. Mallavarapu and G. Marriott, Methods Enzymol., 1998, 291, 63–78 CAS.
- M. B. Smith and J. March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 6th edn, Wiley, Hoboken, New Jersey, 2007 Search PubMed.
- K. B. Sloan and S. A. M. Koch, J. Org. Chem., 1983, 48, 3777–3783 CrossRef CAS.
- J. Hofmann and M. Sernetz, Anal. Biochem., 1983, 131, 180–186 CrossRef CAS.
- T. Kobayashi, Y. Urano, M. Kamiya, T. Ueno, H. Kojima and T. Nagano, J. Am. Chem. Soc., 2007, 129, 6696–6697 CrossRef CAS.
- C. Bueno, M. L. Villegas, S. G. Bertolotti, C. M. Previtali, M. G. Neumann and M. V. Encinas, Photochem. Photobiol., 2002, 76, 385–390 CrossRef CAS.
- J. Hofmann and M. Sernetz, Anal. Chim. Acta, 1984, 163, 67–72 CrossRef CAS.
- M. D. Burke, S. Thompson, R. J. Weaver, C. R. Wolf and R. T. Mayer, Biochem. Pharmacol., 1994, 48, 923–936 CrossRef CAS.
- D. N. Kramer and G. G. Guilbault, Anal. Chem., 1964, 36, 1662–1663 CrossRef CAS.
- T. M. Kitson, Bioorg. Chem., 1996, 24, 331–339 CrossRef CAS.
- We note that the values of kcat/KM for the four AM ethers (4a, 4b, 6, and 9) are (106 ± 40)% those of the analogous acetate esters (2a, 2b, 7, and 10, respectively). The insignificant difference between these values is consistent with the breakdown of the hemiacetal formed initially upon AM-ether hydrolysis being rapid (as expected, see: L. H. Funderburk, L. Aldwin and W. P. Jencks, J. Am. Chem. Soc., 1978, 100, 5444–5459 Search PubMed ) and not limiting the rate of fluorescence generation.
- C. Klein, H.-G. Batz and R. Herrmann, U.S. Patent 6,800,765.
- E. Boonacker, J. Stap, A. Koehler and C. J. F. Van Noorden, Acta Histochem., 2004, 106, 89–96 CrossRef CAS.
- J. Golik, H. S. L. Wong, S. H. Chen, T. W. Doyle, J. J. K. Wright, J. Knipe, W. C. Rose, A. M. Casazza and D. M. Vyas, Bioorg. Med. Chem. Lett., 1996, 6, 1837–1842 CrossRef CAS.
- D. J. Horgan, J. R. Dunstone, J. K. Stoops, E. C. Webb and B. Zerner, Biochemistry, 1969, 8, 2006–2013 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Data on the chemical reactivity (Fig. S1, S5, and S6), long-term stability (Fig. S2 and S4), and enzymatic hydrolysis (Fig. S3) of the profluorophores, and spectral data for novel compounds. See DOI: 10.1039/c0sc00466a |
| This journal is © The Royal Society of Chemistry 2011 |