Triggered structural and property changes in polymeric nanomaterials
Jason M.
Spruell
and
Craig J.
Hawker
*
Materials Research Laboratory, California NanoSystems Institute, Department of Chemistry and Biochemistry, Materials Department, University of California, Santa Barbara, CA, USA. E-mail: hawker@mrl.ucsb.edu; Tel: +1 805 893 7161
First published on 14th October 2010
Abstract
The combination of ordered macromolecular structures and well-defined responsive molecular triggers is enabling precise and amplified structural and property switching in synthetic architectures leading to useful functions. These features are reminiscent of the most sought after designs in nature, specifically the ability of superstructures to form on-demand, store information, perform actuation, and store/release molecular payloads. Such macromolecular systems arise through the precise placement of environmentally responsive elements within polymer chains, an ability only recently possible for synthetic systems. The drivers behind such progress are described and future possibilities for functional macromolecular superstructures discussed.
 Jason M. Spruell | Jason Spruell was born in Nashville, TN and grew up nearby in Chattanooga, TN. He received a BS in chemistry in 2005 from the University of Alabama, performing research with Professors Dave Dixon and Kevin Shaughnessy while there. He completed his PhD in 2009 with Sir Fraser Stoddart, supported by an NSF Graduate Research Fellowship at both UCLA and Northwestern University where he developed efficient protocols to synthesize switchable mechanically interlocked molecules. He recently began postdoctoral work at UC Santa Barbara as an Elings Fellow in Experimental Science with Professor Craig Hawker focused on responsive materials and polymer self-assembly. |
 Craig J. Hawker | Craig J. Hawker, FRS received his BSc (1984) degree from the University of Queensland, Australia his PhD (1988) degree from the University of Cambridge, followed by a post-doctoral fellowship with Professor Jean M.J. Fréchet at Cornell from 1988 to 1990. In 2005 he moved from the IBM Almaden Research Center to the University of California, Santa Barbara where he is a Professor in the Materials, Chemistry and Biochemistry departments and directs the Materials Research Laboratory. His research activities focus on synthetic polymer chemistry, nanotechnology and materials science where he integrates fundamental synthetic studies with the development of nanostructured materials. He holds more than 45 patents, has published over 300 papers. In 2010 he was named as a Fellow of the Royal Society and was recently awarded a 2011 Arthur C. Cope Scholar award from the American Chemical Society. |
Introduction
The convergence of two major areas of polymer science, the synthesis of well-defined macromolecular architectures coupled with the advent of responsive molecular units, is enabling new insights into a grand materials challenge – the production of advanced materials which undergo structural and property changes in response to external stimuli. One needs to look no further than the prevalence of liquid crystalline displays1 to appreciate the applications and functionality achieved through the union of order and triggered movement within assembled molecular structures. In this example, electric field stimulated structural changes bring about useful optical polarization transitions; spawning a multi-billion dollar industry. The main lessons learned are that (1) molecularly defined events provide specificity in both trigger and response which, when ordered into periodically arrayed structures, lead to (2) amplified and cooperative materials responses. From a broader perspective, these technologies still seem primitive when compared to the hierarchy of functional and adaptive macromolecular machinery present in living systems.2,3 In drawing inspiration from these natural examples, which combine multiple triggerable components to achieve a synergistic multi-responsiveness, exploratory studies have recently begun to unravel the challenge of designed responsive materials.4–6 An initial focus on triggered structural changes within synthetic systems and across a range of environments and stimuli is developing with recent seminal examples exploiting distinct molecular triggers – rather than bulk swelling phenomenon – to effect structural and physical changes in macromolecular systems. The synthesis and organization of the triggered, responsive macromolecules – from single-chains, through thin films, nanostructures, and finally assemblies within polymer matrices – will be discussed and drivers for future research suggested.A wide range of adaptive polymeric materials have been developed over the last few decades which take advantage of bulk transitions occurring along the backbone of macromolecules. For example, phase transitions (such as Tg and Tm), swelling/deswelling, and conformational changes of polymers have been used to access “smart” functionality, such as adaptive surfaces,7 self-healing materials,8 and drug delivery.6 While these responses are useful, the degree of control in these traditional systems is lacking. The key difference between the examples described above and those involving molecularly defined transitions is that both the responsive (molecular) element and the ordering/scaffolding (polymer) element may be tuned independently to arrive at modularly defined systems (in terms of both trigger and response). This modularity is more akin to the advanced machinery devised by Nature.
The promise of ever more sophisticated and efficient nanoscale machinery, rivaling the utility of natural systems, is defining both fundamental studies and applications in this expanding field. Significantly, one of the growing strengths of polymer science is the synthetic accessibility and modular design of nanostructured materials which permits the selective incorporation of switchable components, allowing access to dynamic and responsive synthetic systems. While still in its infancy, the field of triggerable polymeric nanomaterials shows great promise and relies on the creative union9 of organic synthesis, polymer science, as well as physical organic chemistry to precisely knit together responsive macromolecular building blocks into functional materials. Early examples of smart materials were driven by bulk phase changes, such as the swelling of polymer domains stimulated by various triggers. In contrast, recent advances have been based upon molecular level transitions, resulting in tunable materials structure and properties. For example, polymeric materials incorporating molecularly responsive elements may be tailored in both stimuli and response through the judicious choice of the specific triggering elements. In such cases, polymers may be induced, singly or in assemblies, to affect well-defined structural and/or property changes in response to a myriad of specific stimuli – the final goal being the design of responsive materials for new information storage, mechanical actuation, self-healing, and molecular storage/release applications (Fig. 1). Arising from such research are materials which exhibit cooperative and amplified responses to multiple, orthogonal triggers. Of even greater significance are the fundamental design principles being developed to achieve responsive polymeric nanostructures and the associated impact of these materials on a wide spectrum of scientific and engineering fields.
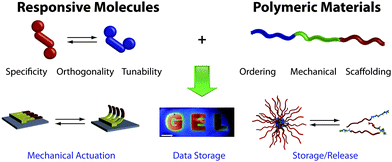 | ||
| Fig. 1 A vision of the combination of responsive molecules and ordering macromolecules. The specificity, orthogonality and tunability of molecular triggers, when mechanically ordered on polymeric scaffolds, enables the production of advanced materials displaying features such as mechanical actuation,13 information storage,33 and storage/release phenomena.41 The promise of the future is grounded in multiresponsive materials capable of functions most often observed in nature. Reproduced in part with permissions from the Nature Publishing Group.13,33 | ||
Ordering of molecular triggers
When discussing order in a chemical context, a variety of definitions may be applied. To a molecular chemist, the constitution of various functional groups held together via covalent bonds, which define a molecule's shape and properties, represents the basis of order. The supramolecular chemist expands this definition further to include the specific assemblies achieved “beyond the molecule” in ensembles of multiple species interacting via directional noncovalent bonds. Polymers too have a specific constitution representing the foundational order built into the structure, for example block copolymers, giving rise to nanoscale morphologies through microphase separation. Supramolecular interactions between single polymer chains, both associative and dissociative, induce self-assembly of appropriately designed macromolecules into discrete ordered domains as enthalpically10,11 or entropically12 stable structures in the bulk and solution. It is this hierarchical structuring, the design for which is programmed through the macromolecular constitution, that empowers the polymer chemist to craft predetermined nanostructures from the bottom-up.Synthetic macromolecules serve as an ideal platform for locating specific molecular receptors or stimuli-responsive molecular elements, oriented along a single-chain or, by virtue of polymer ordering, placed in higher-order nanostructures. Environmentally responsive building blocks provide specificity in both response and trigger and in turn can be tuned through molecular design. For example, azobenzene undergoes a dramatic and reversible shape change in response to photoisomerism of its central double bond. The wavelength of light responsible for this structural change is easily tuned via modification of its molecular structure through the addition of electron donating or withdrawing groups to the azobenzene core. A library (Fig. 2) of other responsive building blocks exist with varied environmental triggers (e.g. changes in light, temperature, pH, solvent, electrochemical state and ion binding), and in select cases orthogonal triggers may be employed within the same architecture to provide multi-responsive materials.13,14 A fundamental and long-term goal for materials chemists is to couple molecularly defined responsive elements with hierarchically ordered polymer nanostructures. Amplification of the molecular response can then be achieved leading to precise switching, both attributes that are most commonly ascribed to natural systems.
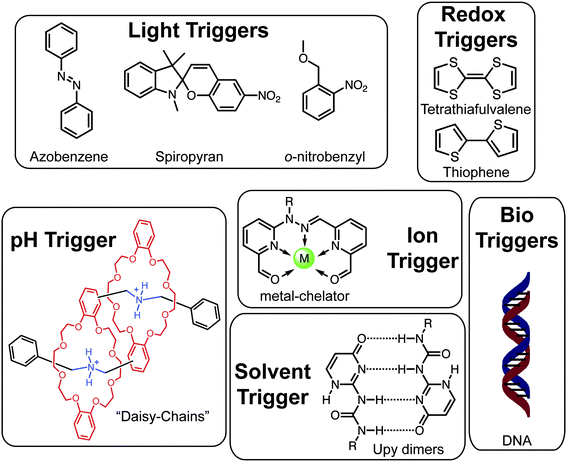 | ||
| Fig. 2 Examples of well-defined molecular triggers responsive to various stimuli. | ||
Molecular changes in single polymer chains
The constitution and dynamics of single polymer chains are of utmost importance in determining polymer behavior. It is therefore necessary to develop means to influence the size, shape, and constitution of single macromolecules. Intensive research has been devoted to the synthesis of well-defined macromolecules through controlled reactions.15–18 It is through these efforts that molecular triggering elements may be incorporated into the polymer chain in a directional fashion. This synthetic accessibility of designer macromolecules is enabling significant advances in the area of polymer dynamics, the most fundamental of which occurs within single polymer chains.While most induced changes of single polymer chains involve shape reorientations occurring along a fixed polymer backbone, macromolecules that adapt their very constitution in response to external stimuli represent an interesting departure from the norm. Lehn and co-workers19,20 recently developed a series of polymer structures in which the monomer units are held together through dynamic covalent bonds. These so called “dynamers” form spontaneously upon mixing bifunctional amine and aldehyde monomer units which condense through imine bonds under slightly acidic conditions. The resulting dynamic libraries of polymers are composed of varying amounts of bifunctional monomer units. As a consequence of their dynamic nature, environmental pressures may directly affect the constitution, and therefore the size and shape, of each polymer within the dynamic library (Fig. 3a). Taking advantage of the combined molecular triggers of donor–acceptor stacking and metal ion binding, dynamic libraries of polymers were precisely ordered into specific constitutions and determined shapes.
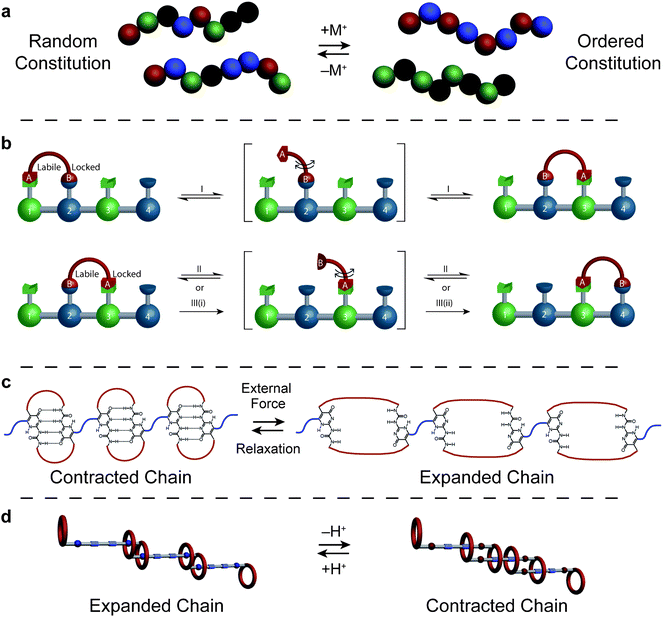 | ||
| Fig. 3 Triggered structural changes within single polymer chains. (a) A dynamic library of exchanging monomers in the form of statistical dynamers are induced to change their constitution, forming ordered dynamers.14 (b) A four base pair synthetic linear motor in which a walker is powered along a track using two orthogonal triggers represented by three distinct inputs (acid, base, neutral).21 (c) Single chains contracted through strong quadruple hydrogen bonding are forced to reversibly expand in response to external force.23 (d) Doubly-threaded rotaxane daisy chain polymers may be induced to expand and contract on demand in response to pH changes.25,26 Reproduced in part with permissions from the Nature Publishing Group.21 | ||
Dynamic covalent bonding was also put to good use within a synthetic linear motor prepared by Leigh and co-workers.21 The researchers devised a track and walker system (Fig. 3b) in which two different orthogonal dynamic covalent bonds – hydrazone and disulfide linkages – were used as “chemical feet” to power the walker molecule unidirectionally across a four “base” pair track. The fine molecular control arising from the dynamic bonds used in its construction as well as the exclusive acid/base sensitivities of each allowed the finely controlled motion of the linear motor. The “passing-leg” gate employed by the walker unit to traverse a unidirectional path is very reminiscent of naturally occurring cargo transport systems comprised of bipedal motor proteins such as dynein and myosin traveling along actin filament tracks powered by adenosine triphosphate hydrolysis.22 The synthetic analogue prepared by the researchers in Scotland is an important example of triggered constitutional switching; moreover, the fine control offered through the orthogonal molecular handles and the prospect of preparing longer polymeric tracks for one or more walkers could lead to the eventual preparation and use of synthetic motor systems for cargo transport as well as the operation of chemical systems far from equilibrium.
Beyond determining macromolecular constitution, discrete molecular triggers may also dictate the shape and stiffness of polymer chains. For example, in more traditional covalent macromolecules reversible stretching of single chains was accomplished by Guan and co-workers23,24 through the incorporation of strongly binding complementary pairs of quadruple hydrogen bonding receptors into every monomer unit along a polymer chain. The strong hydrogen bonds, acting within single chains,23 absorbed stress efficiently through stepwise breaking in preference to the cleavage of covalent bonds also knitting the structure together (Fig. 3c). These noncovalent bonds also form supramolecular crosslinks within entangled films of such macromolecules resulting24 in a rare combination of mechanical properties most usually found in natural polypeptides such as titin, including high toughness with simultaneous elasticity, thus exhibiting shape memory and self-healing properties.
While passive polymer elongation is useful for shock absorption, the incorporation of active functionalities into a polymer chain has enabled the on-demand triggered expansion and contraction of single polymer chains. Stoddart25 and subsequently Grubbs26 recently reported linear polymers composed of doubly-interlocking “daisy chain” monomer units in which pH controlled expansion-contraction behavior was observed. The monomers in each case were composed of dibenzyl ammonium units threaded through crown ether functionalities in such a fashion as to form doubly-interlocking architectures. Following the template-directed synthesis27,28 and stoppering of each monomer unit, condensation polymerization afforded linear polymers held together via interlocking mechanical bonds. The crown ether/ammonium hydrogen bond was easily disrupted and even repulsed following the reversible deprotonation of the ammonium unit, an act which resulted (Fig. 3d) in the contraction (and subsequent expansion) of the linear single chain macromolecules in solution. From these studies, it is clear that flexible components are necessary for single-chain polymer expansion or contraction. Taking advantage of flexible calix[4]arene hinges linking oligothiophene units, Swager and co-workers29 utilized redox-controlled radical π-dimerization between adjacent thiophene domains to modulate the single-chain contraction and expansion of oligothiophene-calix[4]arene hybrid structures. Significantly, these approaches allow the controlled contraction of polymer chains along their linear backbone, though only with a modest percentage of contraction. Future studies focusing on macromolecular structures bearing both hard and soft domains may eventually afford tough, responsive materials displaying large contraction profiles, akin to biological muscle fibers. Finally, a macromolecular system enabling a measured response along the backbone would provide the first evidence of analog contraction (i.e. a dial-able molecular ruler) which would open up new areas of materials design.
Structural changes in the bulk
On progressing from single chains to bulk systems, hierarchically ordered nanostructures become accessible and it is through these structures that significant possibilities arise for tuning both the structure and properties of functional materials. By combining triggered molecular events, amplification can be achieved allowing for macroscopic and useful structural work to occur in response to multiple minor energy inputs. Both ordering and molecular based changes are critical for achieving selective addressability and triggered coherence in these multi-responsive materials. The use of molecularly defined triggering units is potentially more desirable for achieving tunable responsive materials, however, synthetic limitations have constrained past research which has been devoted to a thermoresponsive polymer swelling phenomenon in the bulk, almost exclusively involving poly(N-isopropylacrylamide) (polyNIPAM) as the responsive macromolecule.30 Poly(NIPAM) and its derivatives undergo a thermal phase transition in aqueous environments which results in bulk reversible swelling behavior as well as an inversion in hydrophilicity when cooled below ∼37 °C (a very useful physiological temperature). While these bulk swelling and surface property changes have become the basis for viable technologies such as cell-sheet engineering,31 the utility of the thermoresponsive nature can be significantly enhanced when ordering is introduced. For example, Hayward and co-workers32 recently constructed polyNIPAM crosslinked films over periodic surface topologies that exhibited thermally induced creasing behavior. The creases were used as sequestering domains, the position of which were determined by the original placement of the surface patterns. These areas of sequestration were effectively protected from surface modification during passivation steps, and could be later revealed, following film deswelling, for secondary surface functionalization. The resulting patterned surfaces were used as dynamic scaffolds to host surface-immobilized enzymes as well as adherent cells, both of which could be selectively sequestered and then reversibly exposed following changes in temperature.In order to both decrease the size of surface features while increasing the patterned surface area, ordering induced solely through self-assembly is an important goal with the self-assembly of block copolymers being a critical driver of such designs. These macromolecules consist of two or more distinct polymer blocks joined through a covalent linkage whereby variations in polymer constitution affect the eventual self-assembled structures within bulk phases. The introduction of dynamic and molecularly responsive units within block copolymer phase ordered structures represents a powerful method of achieving useful materials functionality and changes in bulk structure. For example, self-assembly of high molecular weight diblock copolymers composed of equimolar polystyrene (PS) and poly(2-vinyl pyridine) (P2VP) blocks can result in lamellae structures aligned parallel to the surface with a domain size of roughly 50 nm. After chemically transforming the P2VP block into a crosslinked polyelectrolyte, Thomas and co-workers33 observed ion-mediated swelling of the crosslinked block (representing a 1,200% increase in film thickness), leading to tunable photonic gels. As a consequence of the molecular-based swelling process occurring within the highly ordered domains, these gels could switch stop-band position and reflectivity as a function of the ionic strength of the matrix solution as well as the crosslinking density of the P2VP block. These effects were demonstrated visually with written messages contrasted by polymer nanostructures of different crosslinking density, achieving two-color imaging that would appear and disappear upon wetting and drying of the films, respectively. It should be noted that no external fields were necessary in these systems which is a significant advantage.
Whereas self-assembly is efficiently obtained in high molecular weight block copolymers, ordering induced by mesogens in the form of liquid crystalline microdomains also represents an important method for aligning polymeric nanostructures. Moreover, mesogens made up of the photoisomerable chromophore azobenzene allow for specific, photoinduced structural changes. When such azobenzene liquid crystalline domains are crosslinked in polymeric films, the ordering of the liquid crystalline microdomains supply specific directionality to their photoinduced motion within the film. Utilizing these features within a randomly oriented liquid crystalline microdomain structure, Ikeda and co-workers34 applied linearly polarized light at 366 nm to directionally address and photoswitch aligned microdomains of azobenzene liquid crystals within a crosslinked polymer film. Significantly, the overall result of controlled nanoscale incorporation of these phototriggered molecular building blocks is the macroscopic and directed bending of the polymer film spatially defined by the direction of linearly polarized irradiation.
Carrying these concepts further, on-demand alignment of microphase separated block copolymer films was achieved utilizing the molecularly defined interactions of azobenzene photoresponsive elements appended to a polystyrene-b-polymethylacrylate backbone. Through cooperative assembly35 of the azobenzene side-chain moieties under plane-polarized selective irradiation, the orientation of the microphase separated columnar structures bearing azobenzene mesogens could be oriented in any arbitrarily defined direction, specified through the orientation of the applied plane polarized light. Such on-demand control of orientation and morphology through specific molecular responses, represents a powerful paradigm for the fabrication of nanostructured materials.
A central theme in this emerging area is the step-wise addition of more complex building blocks within materials. Such an elegant union of bottom-up and top-down ordering strategies for the alignment of molecularly triggerable units within polymeric nanostructures was recently reported by van Oosten and co-workers.13 These researchers exploited the self-organizing, anisotropic features of liquid crystalline azobenzene mesogens to construct films having an internal gradient of aligned microdomains, thereby dictating the bending direction of the films independent of the light polarization (Fig. 4). Such a gradient of aligned liquid crystalline domains, moving from perpendicularly aligned at the substrate surface to parallel domains at the air interface, formed spontaneously upon the ink-jet printing of mesogenic monomer units in the presence of a surfactant molecule (used to minimize the surface free energy at the substrate interface). The directionality of the aligned phases was dictated by surface templating, created through unidirectional rubbing prior to liquid crystal deposition. Finally, ink-jet printing allowed the deposition of different monomer mixtures, each containing a slightly different azobenzene derivative which photoisomerized at different irradiation wavelengths. When printed and cured in a step-wise fashion, these two different dye-containing liquid crystalline crosslinked films were fabricated into microstructures capable of bending in four modes each differentially induced by irradiation at different wavelengths. Significantly, this behavior was reminiscent of the movement of natural cilia, microstructures also composed of hierarchically assembled macromolecular units, demonstrating initial steps towards mimicking complex natural motor systems.
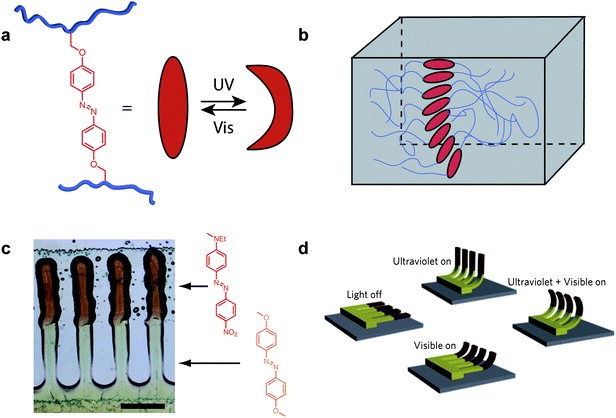 | ||
| Fig. 4 Photo-active artificial cilia.13 (a) Crosslinkable azobenzene moieties were utilized as photoactive mesogens in which a large structural change could be induced through the application of UV light. (b) The mesogens self-aligned upon ink-jet printing onto rubbed surfaces such that a splayed arrangement formed through the thickness of the film; this geometry resulted in differing molecular force and directionality throughout the film thus allowing controlled bending. (c) Artificial cilia printed with two different azobenzene units such that multiresponsive bending was achieved (scale bar = 0.5 mm). (d) A cartoon depicting the four cilia bending modes accessed through the application of different combinations of UV and Visible light. Reproduced in part with permissions from the Nature Publishing Group.13 | ||
Assembling and disassembling polymer nanostructures
By strategically tailoring the backbone of the polymer as well as their side chain groups, molecularly responsive assemblies of macromolecules have become viable candidates for applications ranging from drug delivery to microelectronics.36 The introduction of controlled dynamics to these structures also opens up exciting new applications, such as catch-and-release functionality.37The smallest polymer-based nanoparticles consist of intramolecularly crosslinked single chain macromolecules. Traditionally these systems have been synthesized38 through the covalent crosslinking of single chain macromolecules under dilute conditions to give intramolecularly collapsed polymer nanoparticles. While these systems exhibit interesting physical properties, their non-responsive nature restricts the range of possible applications. In reinvigorating this field and opening up new research opportunities, Meijer and co-workers39 have developed a supramolecular crosslinking strategy built around strong quadruple hydrogen bonding units to reversibly collapse single macromolecules into crosslinked nanoparticles. The particles were formed in dilute solution following the irradiation of o-nitrobenzyl protected 2-ureido-pyrimidinone (UPy) (self-complementary hydrogen bonding units) grafted along the polymer backbone. Photodeprotection of the o-nitrobenzyl group resulted in unhindered hydrogen bonding which, when occurring in dilute solution, resulted in metastable single-chain polymer nanoparticles. Acidification disrupted the hydrogen bonded crosslinks and resulted in the unfolding of the nanoparticles back into coiled single chain macromolecules. Finally, the metastability of these structures was demonstrated by initially processing neat films of the nanoparticles, taking advantage of their high solubility and low viscosity, then thermally triggering the “unfolding” of the single chain nanoparticles to give insoluble supramolecularly crosslinked extended networks – representing a dramatic change in physical and mechanical properties from the starting nanoparticles.
Larger nanostructures made up of multiple polymer chains – micelles and vesicles – represent important vehicles for transporting hydrophobic guests within aqueous environments. Strategies have recently been designed to selectively trigger the formation of such particles under various environmental pressures.40 Amir and co-workers41 utilized the enzymatic cleavage of phosphate moieties appended to one block of a diblock copolymer to induce amphiphilicity, resulting in subsequent polymer self-assembly into colloidal nanostructures (Fig. 5).
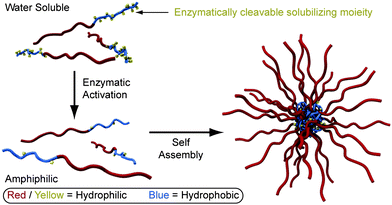 | ||
| Fig. 5 Self-assembling polymer nanostructures.41 Amphiphilicity induced through enzymatic cleavage results in the self-assembly of colloidal nanostructures. | ||
In an orthogonal strategy, Matyjaszewski and co-workers42 reversibly disrupted micelles composed of amphiphilic diblock copolymers utilizing the light-induced isomerization of photochromic spiropyran units appended to the hydrophobic block. Upon irradiation at 365 nm, the spiropyran ring system opens, forming a hydrophilic zwitterionic structure which disrupts the amphiphilic balance of the block copolymer, resulting in destruction of the polymer micelle. Irradiation at 620 nm induced conversion back to the hydrophobic spiropyran ring system, leading to regeneration of the amphiphilic nature and subsequent re-formation of the micellar structures. This hierarchical process which is based on molecular rearrangements leading to changes in the hydrophilic/hydrophobic balance and the assembly/disassembly of nanostructures has the potential to selectively store and release hydrophobic molecular cargo within the nanostructures. Similar behavior was designed into vesicles composed of amphiphilic amide dendrons bearing either photocleavable o-nitrobenzyl protecting groups or photoswitchable azobenzene moieties at the core. Kim and co-workers43 could permanently destroy the vesicular structure and release guest molecules by photoinduced removal of the o-nitrobenzyl protecting group – a triggered event which disrupted the amphiphilic nature of the dendrons. Alternatively, the authors observed that photoirradiation of vesicles bearing azobenzene derivatives at the dendron focal point resulted in temporarily increased permeability of the vesicle walls and the release of guest molecules. These general effects have been combined by Thayumanavan and co-workers,14 where numerous orthogonal triggers were utilized to create a multi-responsive assembling polymer architecture. The combination of temperature sensitive (polyNIPAM), redox sensitive (disulfide linkage), and acid sensitive (acetal) functionalities within a diblock copolymer architecture produced polymeric micelles that would disassemble in response to any one of the specified inputs. A powerful future direction for these types of multiresponsive systems would be the application of logic operations whereby a desired response would only be triggered upon the application of multiple stimuli simultaneously.
In an elegant marriage of small molecule and macromolecule functionality, Sleiman and co-workers37 have produced discrete DNA nanostructures capable of similar responsive assembly/disassembly dynamics. Critical in the formation of these well-ordered DNA assemblies is the presence of rigid, shape-directing small molecule components embedded within single chain DNA oligomers.44 The appropriate choice of base-pairing allows the formation of DNA nanotubes upon hybridization of the structure, where the level of architectural control highlights the power of orthogonal handles. Beyond the controlled self-assembly, Sleiman and co-workers demonstrated that the nanotube compartments could be selectively expanded and contracted through DNA hybridization events triggered through the addition of small complementary single-stranded DNA. These effects were best visualized through the encapsulation and release of gold nanoparticle cargo within the DNA nanotube containers, with a major future goal being DNA/RNA triggered release for precise intra-cell physiological release of therapeutics.
Assemblies within polymer matrices
Beyond performing the dynamic actions themselves, bulk polymer films may also serve as the matrix in which triggered structural ordering takes place. Typically this induced ordering involves the self-assembly of small molecules or nanoparticles within polymer matrices with the polymer matrix playing a critical role, modulating the kinetics and thermodynamics of molecular and mesoscale self-assembly. In practice the polymer film provides a protective and transportable platform in which dynamic processes may take place in functional materials.Ajayaghosh and co-workers45 took advantage of these design criteria to fabricate polystyrene xerogels incorporating supramolecular assemblies of oligo(p-phenylenevinylene) (OPV). These highly fluorescent oligomers are quenched when assembled into well ordered nanostructures due to excited-state energy migration. The polystyrene matrix limits movement of the OPV molecules such that environmental influences determine their assembly behavior. For example, locally heating films containing self-assembled OPV units resulted in their disordering within the matrix with a subsequent increase in fluorescence. These fluorescent images could be erased following solvent annealing of the polystyrene films, an act which softened the polymer matrix and allowed the self-quenching OPV assembly to reform within the film. Utilizing this quenching behavior, the researchers prepared films in which fluorescent images could be thermally written and solvent erased repeatedly. Moreover, because the images were fluorescent in nature, they could not be viewed with the naked eye, nor copied using traditional photocopying technology, suggesting possible document applications.
Self-erasing imaging media, based on triggered structural changes have also been recently developed by Grzybowski and co-workers.46 In this system, metal nanoparticle aggregates were reversibly formed within an organogel film with their associated color changes upon aggregation constituting a multicolor “ink.” The authors had previously found47 that individual Au and Ag nanoparticles stabilized with azobenzene ligands could be induced to form much larger aggregates, called supraspherical assemblies, upon photoisomerising of the azobenzene ligands. This act induces a dipole moment within the ligand such that the alignment of the surface dipoles between different nanoparticles forces them to pack into structured nanoassemblies. The degree of aggregation depended on the length of irradiation while thermal relaxation of the azobenzene units led to redispersion of the individual metal nanoparticles. By incorporating these azobenzene decorated metal nanoparticles into organogel films composed of syndiotactic poly(methyl methacrylate), the irradiative writing of multicolor reversible images in flexible materials (Fig. 6) was possible.
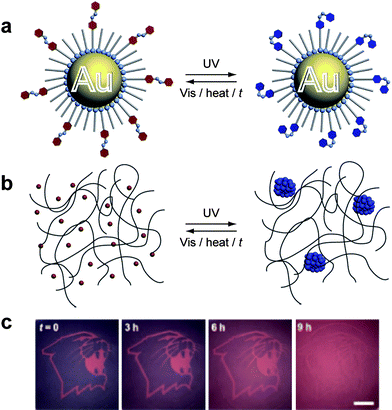 | ||
| Fig. 6 Self-erasing images.46 (a) Gold nanoparticles (NPs) bearing photoactive azobenzene ligands experience reversible surface dipoles in response to UV light. (b) Aggregation assemblies of NPs are formed on-demand within a polymer matrix resulting in macroscopic color changes. (c) Irradiation through a mask writes images on the flexible films that fade over time in visible light; further images may be written on the erased medium. Reproduced in part with permissions from Wiley-VCH.46 | ||
Molecular isomerization also formed the basis for a three-dimensional data storage medium,48 in which holographic images could be written within a bulk polymeric material. Critical to this research was the need to form robust thick films that exhibited little to no shrinkage following the holographic writing event. To achieve these goals, the researchers devised a novel holographic storage strategy based upon the photoisomerization of Dewar benzenes. By crosslinking this responsive unit into thick films at high loadings, changes in the refractive index due to molecular rearrangements were affected in three dimensions upon dual-beam laser irradiation. The quantum amplification of the Dewar benzene rearrangement ensured that nondiffusive strong refractive patterns could be written quickly, while the robust nature of the films produced a high density storage medium with archival shelf-life. By utilizing 1 mm thick films diffraction efficiencies of up to 100%, and M/# of higher than 6 could be achieved with less than 5% degradation in signal after more than 100 days of accelerated testing (cf. greater than 50% signal deterioration for commercial systems after 100 days).
Conclusions and future directions
The focus of recent research in this general area of the design and synthesis of responsive macromolecular systems has been to use a limited number of molecular triggers to perform generally only one function. These simple on/off materials have successfully paved the way to understand the application of molecular force in a cooperative means within ordered ensembles. These results have laid the ground work for the next significant challenge, a systems approach to dynamic materials. Drawing inspiration from the emergent phenomenon observed in systems biology49 and in the fledgling area of systems chemistry,50 the possibilities provided by integration of multiple ordered components and triggering elements is considerable. Correctly combining these elements can result in synergistic feedback mechanisms and cooperativity, eventually enabling complex responses (i.e., reaction cascades within solid state materials leading to amplification, multiple input scenarios leading to logic operations, and maintaining materials in states far from equilibrium). The parallels of this bottom-up assembly of advanced responsive materials with those of the natural world are strong – we have now reached a position where inspiration and mimicry of biological nanostructures are combining.To achieve comparable levels of control and functionality displayed by natural integrated systems, polymer science must show significant progress in the following areas:
1) Transition away from purely thermodynamically stable structures. 51 Specifically, the periodic ordering of responsive elements throughout a bulk material allows the triggered application of force in a directional manner. A challenge in the future of this systems approach will be to develop more robust and orthogonal molecular triggers which can be exploited sequentially or at the same time leading to a number of possible property changes, depending on the sequence of inputs.
2) Design rules for building orthogonality in molecular recognition and switching need to be developed. The endless tunability of DNA sequences illustrates the benefits of orthogonality and further advances in hierarchical ordering – where secondary, tertiary, and subsequent structures are achieved52–54 from modular building units – will also be necessary to not only order responsive elements within the array but to bridge the divide between single component systems and multi-tiered materials (akin to the relationship forged between single cells and higher order tissues demonstrated within natural systems).
3) The introduction of feedback loops will require multiple interacting triggers, where the degree of response and the nature of the response cascade need to be fine-tuned.
4) Further attention must be paid to the precision synthesis of macromolecules through covalent and noncovalent strategies. 55 Developing procedures for controlling the placement of functional groups, for example sequence specific polymers with rational placement of different repeat units along a polymeric backbone are key targets that could benefit from a combined covalent/non-covalent focus.
The success of the initial work described above clearly demonstrates that the cooperative manipulation and ordering of materials on the nanoscale through triggered molecular events is a powerful paradigm in modern materials science and nanotechnology. However, this research area is still in its infancy and as researchers unite small molecule responsive elements with macromolecules in ordered phases, more complex properties, such as regeneration, replication, directed transport, and reaction cascades, traditionally the hallmarks of living systems, may soon be within our synthetic grasp.
Acknowledgements
We thank Peter Allen for the graphical design of Fig. 5. J.M.S. would like to acknowledge the California NanoSystems Institute for an Elings Prize Fellowship in Experimental Science. Financial support from the National Science Foundation (CHE-0957492) and the MRSEC Program (DMR-0520415) is gratefully acknowledged.References
- H. Kawamoto, Proc. IEEE, 2002, 90, 460–500 CrossRef CAS.
- G. Stephanopoulos, H. Alper and J. Moxley, Nat. Biotechnol., 2004, 22, 1261–1267 CrossRef CAS.
- J. Aizenberg, J. C. Weaver, M. S. Thanawala, V. C. Sundar, D. E. Morse and P. Fratzl, Science, 2005, 309, 275–278 CrossRef CAS.
- T. P. Russell, Science, 2002, 297, 964–967 CrossRef CAS.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Prog. Polym. Sci., 2010, 35, 278–301 CrossRef CAS.
- F. Liu and M. W. Urban, Prog. Polym. Sci., 2010, 35, 3–23 CrossRef CAS.
- M. A. Cohen Stuart, W. T. S. Huck, J. Genzer, M. Mueller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. W. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef.
- E. B. Murphy and F. Wudl, Prog. Polym. Sci., 2010, 35, 223–251 CrossRef CAS.
- C. J. Hawker and K. L. Wooley, Science, 2005, 309, 1200–1205 CrossRef CAS.
- C. B. Tang, E. M. Lennon, G. H. Fredrickson, E. J. Kramer and C. J. Hawker, Science, 2008, 322, 429–432 CrossRef CAS.
- M. E. Mackay, A. Tuteja, P. M. Duxbury, C. J. Hawker, B. Van Horn, Z. B. Guan, G. H. Chen and R. S. Krishnan, Science, 2006, 311, 1740–1743 CrossRef CAS.
- S. Gupta, Q. L. Zhang, T. Emrick, A. C. Balazs and T. P. Russell, Nat. Mater., 2006, 5, 229–233 CrossRef CAS.
- C. L. van Oosten, C. W. M. Bastiaansen and D. J. Broer, Nat. Mater., 2009, 8, 677–682 CrossRef CAS.
- A. Klaikherd, C. Nagamani and S. Thayumanavan, J. Am. Chem. Soc., 2009, 131, 4830–4838 CrossRef CAS.
- V. Coessens, T. Pintauer and K. Matyjaszewski, Prog. Polym. Sci., 2001, 26, 337–377 CrossRef CAS.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef.
- S. Perrier and P. Takolpuckdee, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5347–5393 CrossRef CAS.
- C. J. Hawker, Acc. Chem. Res., 1997, 30, 373–382 CrossRef CAS.
- S. Ulrich and J.-M. Lehn, Angew. Chem., Int. Ed., 2008, 47, 2240–2243 CrossRef CAS.
- S. Fujii and J.-M. Lehn, Angew. Chem., Int. Ed., 2009, 48, 7635–7638 CrossRef CAS.
- M. von Delius, E. M. Geertsema and D. A. Leigh, Nat. Chem., 2009, 2, 96–101 Search PubMed.
- S. M. King in Molecular Motors, ed. M. Schliwa, Wiley-VCH, Weinheim, 2003, ch. 2, pp. 45–78 Search PubMed.
- Z. Guan, J. T. Roland, J. Z. Bai, S. X. Ma, T. M. McIntire and M. Nguyen, J. Am. Chem. Soc., 2004, 126, 2058–2065 CrossRef CAS.
- A. M. Kushner, J. D. Vossler, G. A. Williams and Z. Guan, J. Am. Chem. Soc., 2009, 131, 8766–8768 CrossRef CAS.
- L. Fang, M. Hmadeh, J. S. Wu, M. A. Olson, J. M. Spruell, A. Trabolsi, Y. W. Yang, M. Elhabiri, A. M. Albrecht-Gary and J. F. Stoddart, J. Am. Chem. Soc., 2009, 131, 7126–7134 CrossRef CAS.
- P. G. Clark, M. W. Day and R. H. Grubbs, J. Am. Chem. Soc., 2009, 131, 13631–13633 CrossRef CAS.
- D. B. Amabilino and J. F. Stoddart, Chem. Rev., 1995, 95, 2725–2828 CrossRef CAS.
- D. Philp and J. F. Stoddart, Angew. Chem., Int. Ed. Engl., 1996, 35, 1155–1196.
- C. Song and T. M. Swager, Org. Lett., 2008, 10, 3575–3578 CrossRef CAS.
- E. S. Gil and S. A. Hudson, Prog. Polym. Sci., 2004, 29, 1173–1222 CrossRef CAS.
- M. Yamamoto, Y. Akiyama, J. Kobayashi, J. Yang, A. Kikuchi and T. Okano, Prog. Polym. Sci., 2007, 32, 1123–1133 CrossRef CAS.
- J. Kim, J. Yoon and R. C. Hayward, Nat. Mater., 2009, 9, 159–164.
- Y. Kang, J. J. Walish, T. Gorishnyy and E. L. Thomas, Nat. Mater., 2007, 6, 957–960 CrossRef CAS.
- Y. L. Yu, M. Nakano and T. Ikeda, Nature, 2003, 425, 145 CrossRef CAS.
- Y. Morikawa, T. Kondo, S. Nagano and T. Seki, Chem. Mater., 2007, 19, 1540–1542 CrossRef CAS.
- Y. Wang, H. Xu and X. Zhang, Adv. Mater., 2009, 21, 2849–2864 CrossRef CAS.
- P. K. Lo, P. Karam, F. A. Aldaye, C. K. McLaughlin, G. D. Hamblin, G. Cosa and H. F. Sleiman, Nature Chem., 2010, 2, 319–328 CrossRef CAS.
- J. B. Beck, K. L. Killops, T. Kang, K. Sivanandan, A. Bayles, M. E. Mackay, K. L. Wooley and C. J. Hawker, Macromolecules, 2009, 42, 5629–5635 CrossRef CAS.
- E. J. Foster, E. B. Berda and E. W. Meijer, J. Am. Chem. Soc., 2009, 131, 6964–6966 CrossRef CAS.
- Y. Wang, H. Xu and X. Zhang, Adv. Mater., 2009, 21, 2849–2864 CrossRef CAS.
- R. J. Amir, S. Zhong, D. J. Pochan and C. J. Hawker, J. Am. Chem. Soc., 2009, 131, 13949–13951 CrossRef CAS.
- H. I. Lee, W. Wu, J. K. Oh, L. Mueller, G. Sherwood, L. Peteanu, T. Kowalewski and K. Matyjaszewski, Angew. Chem., Int. Ed., 2007, 46, 2453–2457 CrossRef CAS.
- C. Park, J. Lim, M. Yun and C. Kim, Angew. Chem., Int. Ed., 2008, 47, 2959–2963 CrossRef CAS.
- F. A. Aldaye, A. L. Palmer and H. F. Sleiman, Science, 2008, 321, 1795–1799 CrossRef CAS.
- S. Srinivasan, P. A. Babu, S. Mahesh and A. Ajayaghosh, J. Am. Chem. Soc., 2009, 131, 15122–15123 CrossRef CAS.
- R. Klajn, P. J. Wesson, K. J. M. Bishop and B. A. Grzybowski, Angew. Chem., Int. Ed., 2009, 48, 7035–7039 CrossRef CAS.
- R. Klajn, K. J. M. Bishop, M. Fialkowski, M. Paszewski, C. J. Campbell, T. P. Gray and B. A. Grzybowski, Science, 2007, 316, 261–264 CrossRef CAS.
- A. Khan, G. D. Stucky and C. J. Hawker, Adv. Mater., 2008, 20, 3937–3941 CrossRef CAS.
- H. Kitano, Science, 2002, 295, 1662–1664 CrossRef CAS.
- R. F. Ludlow and S. Otto, Chem. Soc. Rev., 2008, 37, 101–108 RSC.
- J. D. Halley and D. A. Winkler, Complexity, 2008, 14, 10–17 CrossRef.
- H. G. Cui, Z. Y. Chen, S. Zhong, K. L. Wooley and D. J. Pochan, Science, 2007, 317, 647–650 CrossRef CAS.
- H. M. Keizer and R. P. Sijbesma, Chem. Soc. Rev., 2005, 34, 226–234 RSC.
- S. H. Yu and H. Colfen, J. Mater. Chem., 2004, 14, 2124–2147 RSC.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |