Accelerated bimolecular reactions in microdroplets studied by desorption electrospray ionization mass spectrometry†
Marion
Girod
a,
Encarnacion
Moyano
b,
Dahlia I.
Campbell
a and
R. Graham
Cooks
*a
aDepartment of Chemistry and Center for Analytical Instrumentation Development, Purdue University, West Lafayette, IN 47907, USA. E-mail: cooks@purdue.edu; Fax: +(765) 494-9421; Tel: +(765) 494-5262
bAnalytical Chemistry Department, University of Barcelona, 08028, Barcelona, Spain
First published on 6th December 2010
Abstract
Functional group derivatization reactions occur in the course of microdroplet/surface collisions in the ambient ionization process of desorption electrospray ionization (DESI). The unique environment in the microdroplet causes rate enhancements of as much as several orders of magnitude in typical bimolecular reactions that proceed through either cationic or anionic intermediates. The environment in the evaporating charged microdroplet differs from that of the bulk: (i) the pH of the solution moves towards the extremes, (ii) the concentrations of the reagents increase, (iii) the relative surface area increases and (iv) collision frequencies increase. The rates of acid-catalyzed reactions, such as the reaction of Girard T reagent with ketosteroids, increase with decreasing pH in positively-charged microdroplets compared to the bulk solution rates. Similarly, the increased pH in evaporating negatively-charged microdroplets contributes to an increase in the rates of base-catalyzed Michael reactions over those recorded under bulk solution conditions. The amount of product formed depends on the reaction time and the droplet size. Nanoelectrospray ionization generates larger droplets than the secondary droplets of DESI so it does not show significant product formation in the analysis period and can be used to analyze products of the DESI experiments. When secondary microdroplets (ca. 1 micron diameter) are generated either by spraying a homogeneous solution of both reagents against an inert surface (reactive DESI) or when a solution of Girard T reagent is sprayed against a solid surface bearing the ketosteroid significant amounts of product are generated. In the case of the Michael reaction with cinnamic acid an alternative dehydrogenated reaction product is formed under microdroplet conditions. Some parallels between the phenomenon reported here and the rate acceleration seen in sonochemistry are noted. The potential value of mass spectrometry in establishing conditions that enhance reaction rates is also indicated. It is possible that these observations will assist in the selection of reaction conditions involving the use of charged microdroplets to enhance the rates of ordinary bulk chemical reactions, especially those involving strong steric hindrance.
Introduction
Over the past few decades increased attention has been given to the fundamental kinetics and mechanisms of atmospheric, biological and other chemical reactions occurring on and within liquid microdroplets. The chemistry of life takes place in small enclosed volumes—cells1—and on a larger scale in atmospheric aerosols,2–4 yet we have only begun to understand and harness the special properties associated with such molecular confinement. In laboratory work, chemical reactions within droplets have mainly been performed by using droplet-based microfluidic systems.5 These systems compartmentalize reactions in droplets of femtoliter to microliter volumes which then act as microreactors.6,7 Compared to conventional bulk synthesis strategies, microfluidic systems allow more precise control of reaction conditions. Compartmentalization in droplets also provides rapid mixing of reagents, allows control of the reaction time, temperature and interfacial properties, and allows ready transport of reagents and products. Droplet-based microfluidics has been used for biochemical screening,8protein crystallization,9,10 enzymatic kinetics11–13 and synthesis of nanoparticles.14–16 The microdroplet environment imposes an increase relative to the bulk solution in the gradients of physical properties (such as temperature, concentration, pressure and density), and this influences the reaction outcome. As one consequence, mass and heat transport in microreactors are much faster than in larger reaction vessels. Microreactors can also be generated using ionic liquids in open air17,18 or water droplets in liquid media without the need for microchannel networks.19Lipid vesicles have been used to create ultra-small volumes encapsulating biological molecules.20 This technique is especially suited for applications aimed at understanding the dynamics of phospholipids membranes or the biological reaction environments inside cells.21–23 Droplets created by emulsion are highly heterogeneous but those formed using microfluidic techniques are generally monodisperse.Once formed, droplets can be manipulated using a wide range of physical mechanisms such as pressure, electric fields,19,24 magnetic fields,25 optical fields26,27 or thermal gradients.28,29 Reagents can be incorporated directly into microdroplets or they can be admixed for example by droplet fusion.19 Levitation of charged microdroplets in electric fields followed by field-induced droplet ionization has allowed the study of microdroplet reactions with gas-phase reactants and product analysis by mass spectrometry.30,31 Similarly, gas/aerosol chemical reactions in microdroplets have been studied by Raman spectroscopy32 and the kinetics modelled. Increased reaction rates are observed in some of the above studies, although the dramatic increases in reaction rate of the type observed in the present study have to our knowledge not previously been reported. Perhaps the closest analog to enhanced reactivity in evaporating droplets is to be found in sonochemistry where acoustic cavitation and subsequent (gaseous droplet) collapse sets up strong physical forces that can drive up reaction rates by large factors.33 The present experiments employ a collapsing solution-phase droplet, not a collapsing gas bubble and collapse is associated with evaporative cooling not strong heating.
Charged microdroplets produced in electrospray ionization represents a general approach to generating microvolume reactors. Conveniently, such reacting systems can be interrogated on-line by mass spectrometric analysis of the contents of the microvolume to provide information on solution-phase reaction intermediates and reaction mechanisms. This assumes that the sampling process does not cause further reaction or modify the chemical species present in the original droplet. This method has seen application in mechanistic studies using electrospray ionization34–36 prior to MS analysis.
Desorption electrospray ionization (DESI) is a mass spectrometric method in which charged primary microdroplets of solvent are pneumatically directed at a surface, producing a liquid film into which solids on the surface are extracted. Subsequent primary droplet collisions release the analyte in secondary microdroplets for analysis by a mass spectrometer.37,38 Chemical derivatization of the condensed phase analyte can be performed in the charged microdroplets released from the surface by incorporating an appropriate reagent into the solvent. This experiment, known as reactive desorption electrospray ionization (reactive DESI),39–43 allows better analyte ionization by selective charge labeling of compounds which do not ionize efficiently. These reactions occur on the time scale of the DESI experiment which is typically on the order of a few seconds or less. This study has as its general aim the development of methods of exploring chemical reactions by mass spectrometry. Solution-phase chemical reactions can be studied almost in real time and with high sensitivity using DESI, the observed reactions occurring in the microdroplet domain and not in bulk solution. The specific aim of the study is to characterize the reactive DESI phenomenon seeking conditions which drive bimolecular reactions in microdroplets. The reaction rates in the small droplets produced during DESI are compared to those in the bulk solution.
Results and discussion
Girard reactions of ketones in microdroplets
Reactions of hydrazines, such as Girard T (GT, a quaternary ammonium hydrazine salt, with cation GT+), with carbonyl compounds such as ketosteroids, generate the corresponding charge-labeled hydrazones. These well-known reactions are acid catalyzed but occur slowly under normal solution-phase conditions.44 The effect on reaction rates of the special conditions represented by the microdroplet environment of the DESI experiment is the subject of this study. DESI was used to generate charged microdroplets of GT solution in methanol–water (MeOH–water) acidified with formic acid and the droplet spray was directed at a steroid spot on an inert surface. In a preliminary set of experiments, several cortisone spots were deposited on a printed polytetrafluoroethylene (PTFE) surface and examined by manually moving the DESI MeOH–water–formic acid spray containing Girard T across the surface while recording DESI spectra. Typical data are summarized in Fig. 1 which shows ion chronograms for the protonated reagent cortisone [M+H]+ and for the hydrazone product [MGT]+, as well as mass spectra at two characteristic time points in the experiment. Data for the Girard reagent are not shown because it is continuously sprayed and always present. The ketone group of cortisone reacts with the amino group of GT in the presence of acid to form the hydrazone, the intact cation of which, [MGT]+, is detected at m/z 474. The data show that product formation maximizes some 10–20 s after the maximum signal for protonated cortisone. This is evident from the chronograms and also from the mass spectra which show product formation increasing relative to reagent over time. The short delay in the signals seen in the macroscopic behavior summarized here is not interpreted to mean that product does not form in the course of single droplet collisions. Other data show small amounts of product formation within a second (the experiment's time resolution) after the spray impacts the solid cortisone spot and generates cortisone ions.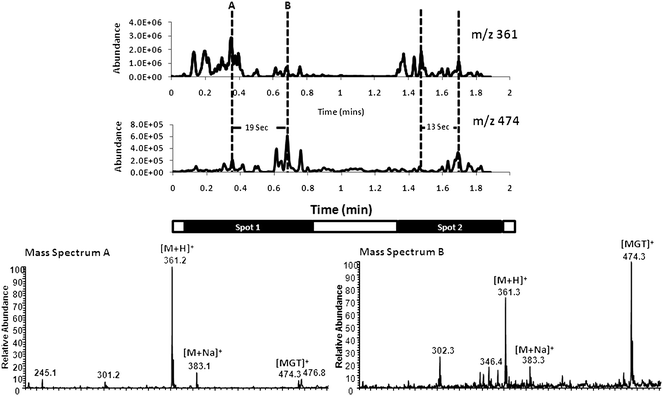 | ||
Fig. 1 Reactive DESI experiment in which a spray of Girard T reagent, 50 ppm in MeOH–water (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20) with 0.5% formic acid, was scanned manually across two separated corticosteroid spots (diameter ca. 2 mm, consisting of 0.2 μg) on a PTFE surface on the seconds time scale indicated by the time bars. Ion chronograms extracted from the mass spectra recorded are shown for the reagent, m/z 361 and the product, m/z 474. Typical mass spectra are shown at 0.3 and ca. 0.6 min. :20) with 0.5% formic acid, was scanned manually across two separated corticosteroid spots (diameter ca. 2 mm, consisting of 0.2 μg) on a PTFE surface on the seconds time scale indicated by the time bars. Ion chronograms extracted from the mass spectra recorded are shown for the reagent, m/z 361 and the product, m/z 474. Typical mass spectra are shown at 0.3 and ca. 0.6 min. | ||
In a second type of DESI experiment, the spray was directed normally at a steroid spot of ca. 2.4 mm diameter on a glass surface and product was allowed to form for 5 min. The surface was then washed and the nanoelectrospray (nanoESI) mass spectrum of the wash was recorded to characterize the products of exposure of the surface to the spray (Fig. 2a). The short time scale of the nanoESI analysis ensures that it does not contribute significantly to product formation. The data again shows reaction product and residual reagent. The cortisone reagent is detected as the protonated molecule [M+H]+ at m/z 361 and as the sodiated adduct [M+Na]+ at m/z 383. The hydrazone ion [MGT]+ is detected at m/z 474 (inset in Fig. 2a).44 These assignments were confirmed by tandem mass spectrometry (MS/MS) experiments and by high resolution MS using an Orbitrap mass spectrometer. The peak at m/z 474.2959 can be accurately assigned as having the exact mass of [MGT]+ measured with an error of −0.6 ppm. Moreover, the MS/MS spectrum of the hydrazone [MGT]+ (Fig. 2b) shows fragmentations characteristic of this species as discussed in the Supplementary Material.† Comparisons made with reactions in bulk solution show that the reaction does not occur to a measurable extent, even on a time scale of minutes as contrasted to DESI. This was established by a control experiment in which nanoESI was used to analyze the bulk mixture of cortisone and GT in aqueous methanol solvent; after 5 min at room temperature it showed no formation of product as indicated by the absence of the peak at m/z 474 (Fig. 2c). Note that rapid product formation appears to be associated with formation of small droplets which are known to be formed in the DESI experiments of Fig. 1 and 2; typical droplet sizes in nanoESI under our conditions are estimated as ca. 5 microns while the secondary droplets on DESI are in the order of 1 micron in diameter.37 Note also that there are features of the experiments that are quite difficult to control and while the behavior described was observed on many occasions, there are cases in which reaction occurs to a smaller extent. (For best results the angle of incidence in the reactive DESI version of the experiment should be somewhat greater than 65°, capillary-to-tip distance must be very small and 50 ppm Girard T solution should be used.)
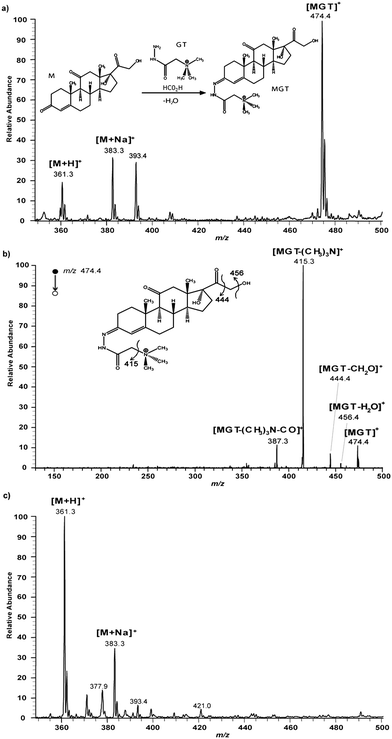 | ||
| Fig. 2 a) Positive ion nanoESI mass spectrum recorded after exposure of 0.15 μg of cortisone as a solid spot of ca. 2.4 mm diameter on glass to microdroplets containing GT reagent for 5 min. Treated surface was washed off in MeOH–water prior to nanoESI. Exposure conditions: GT at 50 ppm in MeOH–water 80:20 (v/v) with 0.5% of formic acid, incident angle to the horizontal: 90°, flow rate: 5 μL min−1, exposure time: 5 min on the spot, tip-to-surface distance: 5 mm, nebulizing gas pressure: 150 psi, high voltage: +1.5 kV. Inset shows the chemical reaction. b) NanoESI-MS/MS spectrum of MGT product detected at m/z 474 after exposure of cortisone to microdroplets containing the GT reagent under the experimental conditions given in a). c) Positive ion nanoESI mass spectrum of a bulk mixture of reagents. Bulk solution: 1.5 μL of cortisone at 100 ppm in MeOH mixed with 25 μL of GT at 50 ppm in MeOH–water 80:20 (v/v) with 0.5% of formic acid, reaction time: 5 min, temperature: 25 °C. | ||
To characterize the reaction more completely, the ratio of abundances of the product ions to the reagent ions was taken as an indication of the reaction yield. Because of the differences in ionization efficiencies between cortisone (M) and the product of reaction of cortisone with GT (MGT), the signal intensity ratio could not be used directly without knowledge of the effect of cortisone and GT on the ionization efficiency of [MGT]. The MGT product, obtained by conventional large scale synthesis, was mixed with a cortisone standard at different known concentrations, in the presence of GT, and the ratios of concentrations plotted against the ratios of the intensities of the corresponding peaks, m/z 474 for [MGT]+ and m/z 361 and m/z 383 for protonated cortisone [M+H]+ (Fig. S2 in the Supplementary Material†). As is the case with the GT reagent, the MGT product has a charge-labeled site and is therefore detected with enhanced intensity in nanoESI compared to neutral cortisone. From this data the ratios of concentrations are actually 2.6 times lower than the observed ratios of intensities. The presence of GT in the analyzed mixture has no significant influence on the calculated reaction yield because the suppression effect is the same for both the MGT product and the cortisone.
The reaction yield (corrected for ionization efficiency) after exposure to the spray of GT was compared with that for bulk solution and also with that when a mixture of the two reagents was simply sprayed against an inert blank surface. Fig. 3 summarizes the ratios of concentrations of MGT product vs.cortisone after reaction under these different experimental conditions. The bulk reaction was done at two different temperatures, in each case for 5 min (Fig. 3a). Although the formation of MGT product is increased by heating, the reaction rate in bulk solution is very low. When a mixture of cortisone and GT is sprayed onto a glass slide and the products then collected and analyzed by nanoESI (Fig. 3b), the derivatization reaction rate increases as reflected in the increased ratio of concentrations MGT/M but a much higher yield is achieved after exposure (5 min) of the cortisone spot to the GT spray (Fig. 3c). These data appear to confirm the greatly enhanced rate of reaction in charged microdroplets. A solution of GT was also sprayed at the cortisone analyte without application of a high voltage for comparison with +1.5 kV high voltage data. The improvement of the reaction yield with high voltage applied to the spray shows a marked charge effect. Even if high voltage is not applied the microdroplets are lightly charged due to disproportionation during the electrospray process45 so some reaction product is observed. Note that the solution contains formic acid so the decrease in pH with solvent evaporation occurs for this reason as well as because of the initial charge on the droplet. An important finding is that the ratio of concentrations (product ions vs. reagent corrected for ionization efficiency) measured for the mixed-and-then-sprayed solution is significantly (2–3 times) lower than that when the cortisone is exposed directly to the charged microdroplets of GT even though the conditions used in the two experiments are otherwise very similar, and literature data indicates that the droplets are of similar size in both experiments. The reasons are not fully understood but they should be related to the one significant difference in the two experiments: the sprayed mixture is homogeneous and the concentrations of M and GT are known and relatively low (100 ppm and 50 ppm, respectively). By contrast the cortisone concentration in DESI is presumably much higher due to the small volume of solvent and the relatively large amount of cortisone on the surface. The process of secondary microdroplet formation in DESI shears off the surface of the liquid film33 and hence creates a droplet composition which might well differ from the equilibrium composition generated by spraying the homogeneous mixture of ketone and Girard reagent.
![Comparison of reaction yields (i.e. ratios of corrected concentrations [(MGT)+/(M + H)+] analyzed by nanoESI for cortisone reaction Girard T for 5 min a) in bulk solution, b) after spraying a mixture of the reagents and c) after exposure of solid cortisone to the GT spray. Bulk conditions: 1.5 μL of cortisone at 100 ppm in MeOH mixed with 25 μL of GT at 50 ppm in MeOH–water 80 : 20 (v/v) with 0.5% of formic acid, reaction time: 5 min, temperature: 25 °C and 50 °C. Mixture spray conditions: 15 μL of the cortisone at 100 ppm in MeOH mixed to 250 μL of the GT solution at 50 ppm in MeOH–water with 0.5% of formic acid, nebulizing gas pressure: 150 psi, flow rate: 5 μL min−1, time: 5 min, high voltages: 0 and +1.5 kV. Solid exposure conditions: GT at 50 ppm in MeOH–water 80 : 20 (v/v) with 0.5% of formic acid, incident angle to the horizontal: 90°, flow rate: 5 μL min−1, exposure time: 5 min across the solid ketone spot of ca. 2.4 mm diameter, tip-to-surface distance: 5 mm, nebulizing gas pressure: 150 psi, high voltage: 0 and +1.5 kV. Ratios of concentrations are calculated from the nanoESI spectra. Bars represent the standard deviation for three replicates.](/image/article/2011/SC/c0sc00416b/c0sc00416b-f3.gif) | ||
| Fig. 3 Comparison of reaction yields (i.e. ratios of corrected concentrations [(MGT)+/(M + H)+] analyzed by nanoESI for cortisone reaction Girard T for 5 min a) in bulk solution, b) after spraying a mixture of the reagents and c) after exposure of solid cortisone to the GT spray. Bulk conditions: 1.5 μL of cortisone at 100 ppm in MeOH mixed with 25 μL of GT at 50 ppm in MeOH–water 80:20 (v/v) with 0.5% of formic acid, reaction time: 5 min, temperature: 25 °C and 50 °C. Mixture spray conditions: 15 μL of the cortisone at 100 ppm in MeOH mixed to 250 μL of the GT solution at 50 ppm in MeOH–water with 0.5% of formic acid, nebulizing gas pressure: 150 psi, flow rate: 5 μL min−1, time: 5 min, high voltages: 0 and +1.5 kV. Solid exposure conditions: GT at 50 ppm in MeOH–water 80:20 (v/v) with 0.5% of formic acid, incident angle to the horizontal: 90°, flow rate: 5 μL min−1, exposure time: 5 min across the solid ketone spot of ca. 2.4 mm diameter, tip-to-surface distance: 5 mm, nebulizing gas pressure: 150 psi, high voltage: 0 and +1.5 kV. Ratios of concentrations are calculated from the nanoESI spectra. Bars represent the standard deviation for three replicates. | ||
Spatial distribution of reaction products
In order to explore further the timing of the reaction and the mechanisms involved in the rate enhancement in DESI-generated microdroplets, the spatial distribution of the compounds after exposure to the spray was examined. Cortisone (0.15 μg) was spotted onto a Teflon surface and the dried spot was exposed for 30 s to a GT spray directed at the center of the spot and normal to the surface. Previous simulations had shown that this procedure allows analyte to dissolve in a thin layer of solution and that subsequently arriving primary droplets splash into this solution to create secondary microdroplets of analyte–reagent mixture.37 The secondary microdroplets rise and then fall back onto the surface in some milliseconds. The resulting surface was subjected to independent DESI analysis using MeOH–water by scanning a DESI probe linearly across the surface and recording mass spectra. The plot shown in Fig. 4a represents the relative concentration of cortisone (M) and cortisone/GT product (MGT) as a function of the position along this scan line across the cortisone spot after exposure to the spray of GT. The results show that some of the cortisone initially present as a uniform spot of 2.6 mm diameter has been converted into product. However, it is clear that product formation does not occur uniformly but only where the splashed microdroplets fall back onto the surface in a ring around the impact site. Both the analyte and the derivatized product are depleted at the point of impact, but the difference in their spatial distributions indicates that a ‘wash out’ effect alone cannot explain the data. Fig. 4b shows the size of the DESI spray (∼1.8 mm in diameter) as measured on water sensitive paper, with an applied voltage of +1.5 kV. The primary droplet impact region is ca. 1.2 mm in diameter and is surrounded by the secondary microdroplet spot.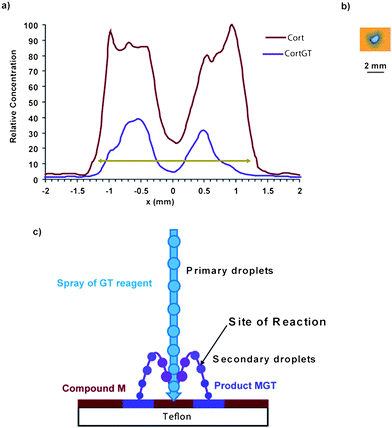 | ||
| Fig. 4 a) Spatial distribution, from the DESI-MS trace, of reagent cortisone M (red) and product MGT (violet) measured after exposure of a spot of cortisone on a Teflon surface (pale green arrow ∼2.4 mm) to GT spray directed at the center of the spot (x = 0). b) Shape and size of the DESI sprayed GT (diameter ∼1.98 mm). Exposure conditions: GT at 50 ppm in MeOH–water 80:20 (v/v) with 0.5% of formic acid, incident angle to the horizontal: 90°, flow rate: 5 μL min−1, exposure time: 30 s, tip-to-surface distance: 5 mm, nebulizing gas pressure: 150 psi, high voltage: +1.5 kV; cortisone: 100 ppm, 1.5 μL spotted on Teflon. DESI analysis conditions: spray: GT at 50 ppm in MeOH–water 80:20 (v/v) with 0.5% of formic acid, incident angle to the horizontal: 50°, flow rate: 3 μL min−1, gas pressure: 150 psi, high voltage: +4.5 kV, velocity: 200 μm s−1. c) Schematic of the overall progress of the reaction in microdroplet environment during DESI. | ||
Previous studies have shown that impacting droplets have average diameters of 2.5–5 μm, while the secondary droplets have average diameters of 0.8–1.9 μm.38 As a result of loss of volume (as pure solvent evaporates) the secondary droplet sizes are reduced during flight, and the pH in the secondary microdroplets is expected to decrease. In fact, the pH value measured at the point of impact is ca. 0.5 instead of 2.0 in the primary droplets, and 3.3 in the bulk solution. Any reaction due to the DESI spray of pure solvent impacting a region containing both cortisone and the Girard reagent is considered negligible based on the results in Fig. 3. While other explanations are possible, the logical one is that reaction occurs on the time scale of the sub-orbital flight of the secondary droplets (≪ 1 s) (Fig. 4c). Because the reaction is acid catalyzed it is driven by the decrease in pH, the increase in concentration, the increase in effective diffusion associated with the droplet evaporation,45Le Chatelier's principle,46 and the effect of confinement discussed further below.21
Fundamental factors controlling droplet reaction rates
In addition to the factors just mentioned, a surface effect will occur when the droplet surface actively favors one or both of the reactants, leading to preferential concentration as a result of polarity or hydrophobicity. This will promote formation of the activated complex and its stabilization at or near the walls of the microdroplet where the surface-to-volume ratio is enhanced in proportion to bulk solution by a factor of 1/r. There are two further considerations: i) the release of energy upon exoergic reaction normally heats a local volume of solution and so tends to reverse the reaction. The opposite occurs for microdroplets which are cooled by evaporation as this removes the heat associated with the reaction so decreasing the rate of the back reaction, ii) there is a smaller temperature rise in small droplets because of more efficient thermal conduction away from the microdroplet.47 We believed that each of these factors may contribute to the enhanced rates observed. The significant difference between the experiments shown in Fig. 3b and 3c suggests that an additional factor is at work in the latter experiment. It is proposed that the secondary microdroplets formed in DESI are highly enriched in the surface layer of the liquid film33 and that these droplets have a higher concentration of hydrophobic cortisone near the droplet surface.Reactions in liquid films and effects of solvent nature
The above explanation of enhanced reaction rates assumes that the reaction indeed takes place within the secondary droplets not in the liquid film created on the surface. The assumption is supported by examination of the influence of the delay time between exposure to the spray and analysis of the reaction yield (Fig. S3, Supplementary Material†). An increase in the delay time from 0.5 to 6 min did not lead to an increase in reaction yield, showing that the reaction stops when no more microdroplets are produced and liquid film reaction is insignificant.Product formation has been studied in a variety of DESI solvents and under various experimental conditions including, electrospray voltages, nebulizing gas pressure and spray tip-to-surface distances (Fig. S4 in the Supplementary Material†). The evaporation process which dictates the size of the droplets is evident in studies of the effects of solvent composition (Fig. S4a in the Supplementary Material†). A higher content of water in the spray solution leads to less efficient evaporation resulting in larger droplets with higher pH's and lower reagent and hence product concentrations (Fig. S5a in the Supplementary Material†). Moreover, water is a product of the reaction and shifts the equilibrium back towards reactants. These results also imply that the kinetics of the reaction in the microdroplet environment are governed by the size of the microdroplets, which dictates the pH, reagent concentrations, surface to bulk ratios and diffusion constants in the microreaction vessel. The effects of the other experimental conditions varied (Fig. S4†) are readily explained by effects on droplet size which can be inferred from measurements of DESI spot sizes. This argument and the data are presented as Supplementary Materials.†
Reaction of a sterically hindered ketone
Studies were carried out on a sterically hindered ketone to test the generality of these findings. The compound chosen, androsterone, is a saturated steroid with a β-methyl group and it shows slow kinetics under normal solution-phase conditions44 (Fig. S6, Supplementary Material†). The reaction product of androsterone with GT was detected (m/z 404.3267, error: −1.0 ppm on the Orbitrap) after exposure to the charged droplets containing the Girard reagent. The rate enhancement for androsterone was even greater than that for cortisone and the reaction yield after exposure was three orders of magnitude higher than that for bulk solution. A schematic of the overall progress of a reaction in the microdroplet environment during DESI was already shown in Fig. 4c, where the secondary microdroplets are proposed to be the sites of the reaction.Reactions involving anionic intermediates
Similar experimental arrangements have also been used to perform solution-phase reactions involving anionic intermediates as typified by the Michael reaction,48 the base-catalyzed addition of the enolate of a ketone or aldehyde to an α,β-unsaturated carbonyl compound. The solvent was doped with dimethyl malonate (DMM) as reagent (Michael donor) and sprayed at a negative voltage onto a methyl cinnamate spot. The nanoESI-MS spectrum obtained after exposure to the spray of DMM at −3.5 kV in methanol–water containing sodium hydroxide is shown in Fig. 5a. The reaction was carried out in the negative ion mode and the analysis was performed in the positive ion mode. Both methyl cinnamate and its product (inset in Fig. 5a) ionize better in the positive ion mode because of the lack of acidic proton and do so with similar ionization efficiencies. These compounds were detected as sodiated adducts [M+Na]+ at m/z 171 and [M(DMM)+Na]+ at m/z 317 (m/z 317.0959, error: −11.3 ppm using the Orbitrap), respectively. The MS/MS product ion spectrum of the sodiated adduct of the product [M(DMM)+Na]+ (Fig. 5b) shows characteristic dissociation behavior with the elimination of methanol (32 Da) and methyl formate (60 Da) from the DMM portion of the product and loss of a methyl acetate molecule (74 Da) from the methyl cinnamate portion, which confirms the structural assignment.![a) Positive ion nanoESI mass spectrum, after blank subtraction, recorded after exposure of 0.15 μg of methyl cinnamate as a solid spot on glass of ca. 2.4 mm diameter to microdroplets containing DMM reagent. Exposure conditions: DMM at 100 ppm in MeOH–water 80 : 20 (v/v) with 200 ppm of sodium hydroxide, incident angle to the horizontal: 90°, flow rate: 5 μL min−1, exposure time: 5 min, tip-to-surface distance: 5 mm, nebulizing gas pressure: 150 psi, high voltage: −3.5 kV. Inset shows the chemical reaction. b) NanoESI-MS/MS spectrum of [M(DMM)+Na]+ product detected at m/z 317 after exposure of methyl cinnamate to microdroplets containing the DMM reagent under the same experimental conditions. c) Positive ion nanoESI mass spectrum of bulk solution. Bulk conditions: 1.5 μL of methyl cinnamate at 100 ppm in MeOH mixed with 25 μL of DMM at 100 ppm in MeOH–water 80 : 20 (v/v) with 200 ppm of sodium hydroxide, reaction time: 5 min, temperature: 25 °C.](/image/article/2011/SC/c0sc00416b/c0sc00416b-f5.gif) | ||
| Fig. 5 a) Positive ion nanoESI mass spectrum, after blank subtraction, recorded after exposure of 0.15 μg of methyl cinnamate as a solid spot on glass of ca. 2.4 mm diameter to microdroplets containing DMM reagent. Exposure conditions: DMM at 100 ppm in MeOH–water 80:20 (v/v) with 200 ppm of sodium hydroxide, incident angle to the horizontal: 90°, flow rate: 5 μL min−1, exposure time: 5 min, tip-to-surface distance: 5 mm, nebulizing gas pressure: 150 psi, high voltage: −3.5 kV. Inset shows the chemical reaction. b) NanoESI-MS/MS spectrum of [M(DMM)+Na]+ product detected at m/z 317 after exposure of methyl cinnamate to microdroplets containing the DMM reagent under the same experimental conditions. c) Positive ion nanoESI mass spectrum of bulk solution. Bulk conditions: 1.5 μL of methyl cinnamate at 100 ppm in MeOH mixed with 25 μL of DMM at 100 ppm in MeOH–water 80:20 (v/v) with 200 ppm of sodium hydroxide, reaction time: 5 min, temperature: 25 °C. | ||
The reaction yield after exposure to the DMM spray has been compared with that for reactions in bulk solution (Fig. 5c) as well as for sprayed mixtures. Fig. 6 shows the ratios of concentrations of the Michael product vs. that of the methyl cinnamate reagent after reaction under different experimental conditions. Although the formation of the M(DMM) product was observed in bulk conditions (Fig. 6a), the ratio of concentrations M(DMM)/M was some 15 times higher when methyl cinnamate was exposed to the negatively-charged microdroplets of DMM (Fig. 6c). An increase in the negative applied voltage produces smaller droplets with higher negative charges due to the electrospray process.38 Thus, the solution becomes more basic and the reagent concentrations increase as evaporation occurs. The pH in the secondary microdroplets was measured as 13.3 (compared to 12.6 for the primary droplets) with an applied voltage of −3.5 kV while the value for the bulk solution was only 10.1. We note that when a positive high voltage was applied to the spray, the pH of the microdroplets was lower (around 11 in the secondary droplets at an applied voltage of +3.5 kV) and the reaction yield was similar to that obtained with the bulk solution. These experiments with a basic-catalyzed reaction provide evidence of the important role of the pH in the enhancement in reaction yield in the microdroplet environment. Importantly, they also implicate the microdroplet size as one basis for the enhanced reaction rates.
![Comparison of the reaction yields (i.e. ratios of concentrations [{M(DMM)}+/(M+H)+] analyzed by nanoESI for the reaction of methyl cinnamate analyte a) in bulk solution, b) after spraying a mixture of the reagents and c) after exposure of the solid ester (0.15 μg) to DMM spray. Bulk conditions: 1.5 μL of cinnamic acid at 100 ppm in MeOH mixed with 25 μL of DMM at 100 ppm and NaOH at 200 ppm in MeOH–water 80 : 20 (v/v), reaction time: 5 min, temperature: 25 °C and 50 °C. Mixed and sprayed conditions: 15 μL of cinnamic acid at 100 ppm in MeOH mixed with 250 μL of DMM at 100 ppm and NaOH at 200 ppm in MeOH–water 80 : 20 (v/v), nebulizing gas pressure: 150 psi, flow rate: 5 μL min−1, time: 5 min, high voltages: 0 and −3.5 kV. Exposure conditions: DMM at 100 ppm in MeOH–water 80 : 20 (v/v) with NaOH 200 ppm, incident angle to the horizontal: 90°, flow rate: 5 μL min−1, exposure time: 5 min across the spot of ca. 2.4 mm diameter, tip-to-surface distance: 5 mm, nebulizing gas pressure: 150 psi, high voltage: 0 and −3.5 kV. Ratios of concentrations are calculated from the nanoESI spectra. Bars represent the standard deviation of analysis for three replicates.](/image/article/2011/SC/c0sc00416b/c0sc00416b-f6.gif) | ||
| Fig. 6 Comparison of the reaction yields (i.e. ratios of concentrations [{M(DMM)}+/(M+H)+] analyzed by nanoESI for the reaction of methyl cinnamate analyte a) in bulk solution, b) after spraying a mixture of the reagents and c) after exposure of the solid ester (0.15 μg) to DMM spray. Bulk conditions: 1.5 μL of cinnamic acid at 100 ppm in MeOH mixed with 25 μL of DMM at 100 ppm and NaOH at 200 ppm in MeOH–water 80:20 (v/v), reaction time: 5 min, temperature: 25 °C and 50 °C. Mixed and sprayed conditions: 15 μL of cinnamic acid at 100 ppm in MeOH mixed with 250 μL of DMM at 100 ppm and NaOH at 200 ppm in MeOH–water 80:20 (v/v), nebulizing gas pressure: 150 psi, flow rate: 5 μL min−1, time: 5 min, high voltages: 0 and −3.5 kV. Exposure conditions: DMM at 100 ppm in MeOH–water 80:20 (v/v) with NaOH 200 ppm, incident angle to the horizontal: 90°, flow rate: 5 μL min−1, exposure time: 5 min across the spot of ca. 2.4 mm diameter, tip-to-surface distance: 5 mm, nebulizing gas pressure: 150 psi, high voltage: 0 and −3.5 kV. Ratios of concentrations are calculated from the nanoESI spectra. Bars represent the standard deviation of analysis for three replicates. | ||
An alternative reaction pathway
Interestingly, when cinnamic acid was used as the analyte, the dehydrogenated form of the product was observed. Indeed, the positive ion mode nanoESI-MS spectrum obtained after exposure of the cinnamic acid spot to the DMM spray at −3.5 kV in methanol–water containing base (Fig. 6a) showed a peak at m/z 301 which can be assigned to the sodiated adduct of the dehydrogenated product [M(DMM)-H2 + Na]+ (m/z 301.0625, error: −18.9 ppm on the Orbitrap), while cinnamic acid was detected as its sodiated adduct at m/z 171. In order to localize the hydrogens involved in this oxidation, H/D exchange experiments were done. By diluting the cinnamic acid in d4-methanol, the hydrogen of the hydroxide group and the hydrogen in β-position were exchanged, forming doubly-deuterated cinnamic acid (d2-M) detected with a 2 Da mass shift at m/z 173 (Fig. 7b and insert). After exposure of the deuterated cinnamic acid to the spray of DMM under the same conditions (no mass shift was observed for m/z 301 suggesting that the d2-cinnamic acid loses, during the reaction with the DMM, a D2 molecule possibly to form the cyclic product shown in the insert. The elimination of the acidic hydrogen of the hydroxide group helps explain the poor ionization efficiency of the product in the negative mode compared to cinnamic acid. The MS/MS spectrum of the sodiated product [M(DMM)-H2 + Na]+ (Fig. 7c) shows characteristic dissociation behavior with the elimination of methanol (32 Da) and methyl formate (60 Da) from the DMM portion and the loss of the C2H2O2 cyclic group (58 Da), which confirms the assignment. By contrast, formation of the M(DMM) product occurs without dehydrogenation under bulk conditions (sodiated adduct detected at m/z 303 in Fig. 7d), indicating that the charges produced in the microdroplet environment induce a specific ionic mechanism which is also favored by the labile acidic proton of the hydroxide group.![a) Positive ion nanoESI mass spectrum, after blank subtraction, recorded after exposure of cinnamic acid (0.15 μg spot of ca. 2.4 mm diameter on glass) to microdroplets containing DMM reagent. Exposure conditions: DMM at 100 ppm in MeOH–water 80 : 20 (v/v) with 200 ppm of sodium hydroxide, incident angle to the horizontal: 90°, flow rate: 5 μL min−1, exposure time: 5 min across the spot, tip-to-surface distance: 5 mm, nebulizing gas pressure: 150 psi, high voltage: −3.5 kV. Inset shows the chemical reaction. b) Positive ion nanoESI mass spectrum recorded after exposure of d2-cinnamic acid (100 ppm on glass slide) to microdroplets containing the DMM reagent in the same experimental conditions. c) nanoESI-MS/MS spectrum of [M(DMM)-H2+Na]+ product detected at m/z 301 after exposure of methyl cinnamate to microdroplets containing the DMM reagent in the same experimental conditions. d) Positive ion nanoESI mass spectrum of the bulk solution. Bulk conditions: 1.5 μL cinnamic acid at 100 ppm in MeOH mixed with 25 μL of DMM at 100 ppm in MeOH–water 80 : 20 (v/v) with 200 ppm of sodium hydroxide, reaction time: 5 min, temperature: 25 °C.](/image/article/2011/SC/c0sc00416b/c0sc00416b-f7.gif) | ||
| Fig. 7 a) Positive ion nanoESI mass spectrum, after blank subtraction, recorded after exposure of cinnamic acid (0.15 μg spot of ca. 2.4 mm diameter on glass) to microdroplets containing DMM reagent. Exposure conditions: DMM at 100 ppm in MeOH–water 80:20 (v/v) with 200 ppm of sodium hydroxide, incident angle to the horizontal: 90°, flow rate: 5 μL min−1, exposure time: 5 min across the spot, tip-to-surface distance: 5 mm, nebulizing gas pressure: 150 psi, high voltage: −3.5 kV. Inset shows the chemical reaction. b) Positive ion nanoESI mass spectrum recorded after exposure of d2-cinnamic acid (100 ppm on glass slide) to microdroplets containing the DMM reagent in the same experimental conditions. c) nanoESI-MS/MS spectrum of [M(DMM)-H2+Na]+ product detected at m/z 301 after exposure of methyl cinnamate to microdroplets containing the DMM reagent in the same experimental conditions. d) Positive ion nanoESI mass spectrum of the bulk solution. Bulk conditions: 1.5 μL cinnamic acid at 100 ppm in MeOH mixed with 25 μL of DMM at 100 ppm in MeOH–water 80:20 (v/v) with 200 ppm of sodium hydroxide, reaction time: 5 min, temperature: 25 °C. | ||
Conclusion
Charged microdroplets containing particular reagents and produced by the methods used in desorption electrospray ionization have interesting properties as reaction vessels. The unique environment in the droplet and/or at the droplet–surface interface causes rate enhancements of as much as several orders of magnitude in simple organic reactions occurring via cationic and anionic intermediates. In such small volumes, the pH of the solution is easily modified by evaporation, the reagent concentrations increase and the contained reagents experience increased numbers of collisions which may also have greater efficiency in product formation compared to collisions in bulk solution. It seems likely that similar phenomena occur with other droplet-based ionization methods, including nanoelectrospray, under appropriate conditions. The conditions used here for nanoESI analysis involve short ionization periods and no product accumulation, so significant reaction is not observed. Droplet surface concentration of the ketone and reagents are suggested as a secondary factor contributing to the rate acceleration.The reasons for the differences between droplets generated by DESI and those generated by spraying a solution containing both reagents may lie in the unusual composition of the droplets generated in DESI. The process gives secondary droplets which preferentially contain surface layers from the liquid film covering the solid being examined by DESI. DESI appears to be an easy way to form ultra-small reaction volumes and to test the reactivity of various chemical systems in this environment. The physical properties of the droplets can be controlled by modifying the spray parameters. The products of the reaction can be directly analyzed on-line by MS. The methodology described here should be applicable to characterizing reaction systems in order to accelerate the rates of formation of particular desired products. In one case different reaction products result from the peculiar conditions of the DESI experiment compared to those that obtain when the same reagents are simply mixed.
Experimental
Experiments were carried out using a commercial Thermo Scientific LTQ (San Jose, CA, USA) linear ion trap mass spectrometer operated in both the positive and negative ion modes. The capillary temperature was at 150 °C, the capillary voltage at 15 V and the tube-lens voltage 65 V. The spray of microdroplets containing the reagent was produced using a custom-built, automated DESI source fitted with a 100 μm diameter capillary which was used in all experiments. The home-built DESI ion source is similar to the OmniSpray source from Prosolia Inc. (Indianapolis, IN, USA). Nitrogen gas was used as the nebulizing gas. Nanoelectrospray data were recorded using a drawn glass capillary (tip diameter of 10 μm) as the nanospray emitter source using a typical flow rate of 20 nL min−1.Exact mass measurements were performed using a commercial Thermo Scientific LTQ-Orbitrap XL mass spectrometer with a resolution set to 100,000 and using cortisone or methyl cinnamate protonated and sodiated adducts as internal standards for the calibration. Product ion mass spectra were recorded using an isolation width of 1.5 Th, a normalized collision energy of 20–40% and a Mathieu parameter, qz, of 0.25.
All the pHs were measured with a highly sensitive pH paper (pHydrionUltrafine, Micro Essential Laboratory, Brooklyn, NY, USA). For the measurement of the pH in the primary droplets, the pH paper was exposed to the spray of droplets for 15 s with a tip-to-surface distance of 5 mm and the pH was indicated by the colors in the center of the impact spot. The pH paper was placed 0.7 mm away from the horizontal axis of the sprayer tip, for 15 s and with a tip-to-surface distance of 5 mm, in order to measure the pH of the secondary microdroplets. All chemicals were purchased from Sigma-Aldrich (Milwaukee, WI, USA).
Reaction with steroids
In the preliminary experiments, standard DESI conditions were used to examine cortisone spotted onto a PTFE surface. Analyte solution (1.5 μL at 100 ppm in methanol) was spotted on the surface, allowed to dry and exposed to the spray of Girard T reagent (GT) at 50 ppm in methanol–water (MeOH–water) 80:20 (v/v) containing 0.5% of formic acid, with an incident angle to the horizontal of 65° and at a constant flow rate of 5 μL min−1.
In subsequent experiments, the steroid reagent was dissolved (1.5 μL at 100 ppm in methanol) spotted on the surface, allowed to dry and exposed to the spray of Girard T reagent (GT) at 50 ppm in MeOH–water 80:20 (v/v) containing 0.5% of formic acid, with an incident angle to the horizontal of 90° and at a constant flow rate of 5 μL min−1. For the comparison of the reaction yield with the reaction in bulk (Fig. 2c), the steroid was spotted on a glass slide and the GT reagent was sprayed onto the whole spot for 5 min, with a tip-to-surface distance of 5 mm, a nebulizing gas pressure of 150 psi and two different voltages (0 and +1.5 kV). After exposure, the products present on the glass surface were dissolved in 2 μL of MeOH–water 80:20 (v/v), collected and analyzed by nanoESI using a nanospray voltage of +1.8 kV.
In order to obtain the images of the spot after exposure and for the study of the influence of the spray parameters (Fig. 4 and 5), the compounds were spotted on printed rough Teflon and the GT reagent was sprayed onto the center of the spot for 30 s, with different tip-to-surface distances, nebulizing gas pressures, high voltages and solvent compositions. After exposure, the products present on the surface were analyzed by DESI using GT at 50 ppm in MeOH–water 80:20 (v/v) with 0.5% of formic acid as the solvent spray with an incident angle of 50°, at a constant flow rate of 3 μL min−1 and with an applied voltage of +4.5 kV. The use of printed rough Teflon substrate allows a better desorption efficiency of the products during the DESI analysis compared to glass substrate.
For the sprayed mixtures experiments (Fig. 3b), 15 μL of the steroid solution at a concentration of 100 ppm in MeOH were mixed with 250 μL of the GT solution at 50 ppm in MeOH–water containing 0.5% of formic acid. The mixture of compounds was then sprayed onto a glass slide using a nebulizing gas pressure of 150 psi, at a constant flow rate of 5μL min−1 and at two different voltages (0 and +1.5 kV) over a period of 5 min. The products were collected and analyzed by nanoESI with a nanospray voltage of +1.8 kV, while the mixture solutions were directly analyzed by nanoESI after 5 min at different temperatures and solvent compositions for the bulk experiments (Fig. 3a).
Michael reaction
Analyte solution (1.5 μL at 100 ppm in methanol) was spotted on the surface, allowed to dry and exposed to the spray of dimethyl malonate (DMM) at a concentration of 100 ppm in methanol–water (MeOH–water) 80:20 (v/v) containing 200 ppm of sodium hydroxide with a negative applied high voltage. The H/D exchange experiments were done by diluting the cinnamic acid in CD3OD (d4-methanol). The same procedure as for the reaction with GT has been used for the studies of the different experimental conditions.
Note added in proof: Since submission of this work we have learned of related experiments by Zare and co-workers.49
Acknowledgements
This work was supported by the National Science Foundation (NSF0848650) and the Office of Naval Research (N00014-05-1-0454). E. Moyano is grateful to “Generalitat de Catalunya” for the research grant (BE-DGR, 2008-BE2-000159).References
- M. J. Waldner and M. F. Neurath, Curr. Gene Ther., 2009, 9, 239–247 Search PubMed.
- M. D. King, A. R. Rennie, C. Pfrang, A. V. Hughes and K. C. Thompson, Atmos. Environ., 2010, 44, 1822–1825 CrossRef CAS.
- V. Michaud, I. El Haddad, Y. Liu, K. Sellegri, P. Laj, P. Villani, D. Picard, N. Marchand and A. Monod, Atmos. Chem. Phys., 2009, 9, 5119–5130 CAS.
- Z. Juranyi, M. Gysel, J. Duplissy, E. Weingartner, T. Tritscher, J. Dommen, S. Henning, M. Ziese, A. Kiselev, F. Stratmann, I. George and U. Baltensperger, Phys. Chem. Chem. Phys., 2009, 11, 8091–8097 RSC.
- H. Song, D. L. Chen and R. F. Ismagilov, Angew. Chem., Int. Ed., 2006, 45, 7336–7356 CrossRef CAS.
- M. Brivio, W. Verboom and D. N. Reinhoudt, Lab Chip, 2006, 6, 329–344 RSC.
- Q. Q. Zhang, S. J. Zeng, J. H. Qin and B. C. Lin, Electrophoresis, 2009, 30, 3181–3188 CrossRef CAS.
- L. M. Fidalgo, G. Whyte, B. T. Ruotolo, J. L. P. Benesch, F. Stengel, C. Abell, C. V. Robinson and W. T. S. Huck, Angew. Chem., Int. Ed., 2009, 48, 3665–3668 CrossRef.
- B. Zheng, J. D. Tice, L. S. Roach and R. F. Ismagilov, Angew. Chem., Int. Ed., 2004, 43, 2508–2511 CrossRef CAS.
- B. Zheng, L. S. Roach and R. F. Ismagilov, J. Am. Chem. Soc., 2003, 125, 11170–11171 CrossRef CAS.
- H. N. Joensson, M. L. Samuels, E. R. Brouzes, M. Medkova, M. Uhlen, D. R. Link and H. Andersson-Svahn, Angew. Chem., Int. Ed., 2009, 48, 2518–2521 CrossRef CAS.
- H. Song and R. F. Ismagilov, J. Am. Chem. Soc., 2003, 125, 14613–14619 CrossRef CAS.
- Z. Y. Han, W. T. Li, Y. Y. Huang and B. Zheng, Anal. Chem., 2009, 81, 5840–5845 CrossRef CAS.
- L. Frenz, A. El Harrak, M. Pauly, S. Begin-Colin, A. D. Griffiths and J. C. Baret, Angew. Chem., Int. Ed., 2008, 47, 6817–6820 CrossRef CAS.
- Y. Schaerli, R. C. Wootton, T. Robinson, V. Stein, C. Dunsby, M. A. A. Neil, P. M. W. French, A. J. deMello, C. Abell and F. Hollfelder, Anal. Chem., 2009, 81, 302–306 CrossRef CAS.
- I. Shestopalov, J. D. Tice and R. F. Ismagilov, Lab Chip, 2004, 4, 316–321 RSC.
- P. Dubois, G. Marchand, Y. Fouillet, J. Berthier, T. Douki, F. Hassine, S. Gmouh and M. Vaultier, Anal. Chem., 2006, 78, 4909–4917 CrossRef CAS.
- M. Lopez-Pastor, A. Dominguez-Vidal, M. J. Ayora-Canada, T. Laurell, M. Valcarcel and B. Lendl, Lab Chip, 2007, 7, 126–132 RSC.
- T. Taniguchi, T. Torii and T. Higuchi, Lab Chip, 2002, 2, 19–23 RSC.
- D. T. Chiu, C. F. Wilson, F. Ryttsen, A. Stromberg, C. Farre, A. Karlsson, S. Nordholm, A. Gaggar, B. P. Modi, A. Moscho, R. A. Garza-Lopez, O. Orwar and R. N. Zare, Science, 1999, 283, 1892–1895 CrossRef CAS.
- D. T. Chiu, R. M. Lorenz and G. D. M. Jeffries, Anal. Chem., 2009, 81, 5111–5118 CrossRef CAS.
- R. Karlsson, A. Karlsson, A. Ewing, P. Dommersnes, J. F. Joanny, A. Jesorka and O. Orwar, Anal. Chem., 2006, 78, 5960–5968 CAS.
- T. Sunami, K. Hosoda, H. Suzuki, T. Matsuura and T. Yomo, Langmuir, 2010, 26, 8544–8551 CrossRef CAS.
- T. P. Hunt, D. Issadore and R. M. Westervelt, Lab Chip, 2008, 8, 81–87 RSC.
- N. T. Nguyen, K. M. Ng and X. Y. Huang, Applied Physics Letters, 2006, 89.
- G. D. M. Jeffries, J. S. Kuo and D. T. Chiu, J. Phys. Chem. B, 2007, 111, 2806–2812 CrossRef CAS.
- D. T. Chiu and R. M. Lorenz, Acc. Chem. Res., 2009, 42, 649–658 CrossRef CAS.
- M. L. Cordero, D. R. Burnham, C. N. Baroud and D. McGloin, Applied Physics Letters, 2008, 93.
- K. T. Kotz, K. A. Noble and G. W. Faris, Appl. Phys. Lett., 2004, 85, 2658–2660 CrossRef CAS.
- R. L. Grimm, R. Hodyss and J. L. Beauchamp, Anal. Chem., 2006, 78, 3800–3806 CrossRef CAS.
- H. I. Kim, H. Kim, Y. S. Shin, L. W. Beegle, S. S. Jang, E. L. Neidholdt, W. A. Goddard, J. R. Heath, I. Kanik and J. L. Beauchamp, J. Am. Chem. Soc., 2010, 132, 2254–2263 CrossRef CAS.
- A. K. Y. Lee and C. K. Chan, Atmos. Environ., 2007, 41, 4611–4621 CrossRef CAS.
- K. S. Suslick, Science, 1990, 247, 1439–1445 CrossRef CAS.
- L. S. Santos, G. B. Rosso, R. A. Pilli and M. N. Eberlin, J. Org. Chem., 2007, 72, 5809–5812 CrossRef CAS.
- J. Griep-Raming, S. Meyer, T. Bruhn and J. O. Metzger, Angew. Chem., Int. Ed., 2002, 41, 2738–2742 CrossRef CAS.
- M. N. Eberlin, Eur. J. Mass Spectrom., 2007, 13, 19–28 CrossRef CAS.
- A. B. Costa and R. G. Cooks, Chem. Phys. Lett., 2008, 464, 1–8 CrossRef CAS.
- A. Venter, P. E. Sojka and R. G. Cooks, Anal. Chem., 2006, 78, 8549–8555 CrossRef CAS.
- J. E. Barbara, J. R. Eyler and D. H. Powell, Rapid Commun. Mass Spectrom., 2008, 22, 4121–4128 CrossRef CAS.
- G. Huang, H. Chen, X. Zhang, R. G. Cooks and Z. Ouyang, Anal. Chem., 2007, 79, 8327–8332 CrossRef CAS.
- L. Nyadong, E. G. Hohenstein, A. Galhena, A. L. Lane, J. Kubanek, C. D. Sherrill and F. M. Fernandez, Anal. Bioanal. Chem., 2009, 394, 245–254 CrossRef CAS.
- Y. Song and R. G. Cooks, J. Mass Spectrom., 2007, 42, 1086–1092 CrossRef.
- R. Sparrapan, L. S. Eberlin, R. Haddad, R. G. Cooks, M. N. Eberlin and R. Augusti, J. Mass Spectrom., 2006, 41, 1242–1246 CrossRef CAS.
- O. H. Wheeler, J. Chem. Educ., 1968, 45, 435–437 CrossRef CAS.
- J. V. Iribarne and B. A. Thomson, J. Chem. Phys., 1976, 64, 2287–2294 CrossRef CAS.
- M. Planck, Ann. Phys., 1934, 411, 759–768 CrossRef.
- G. D. M. Jeffries, J. S. Kuo and D. T. Chiu, Angew. Chem., Int. Ed., 2007, 46, 1326–1328 CrossRef CAS.
- T. Tokoroyama, Eur. J. Org. Chem., 2010, 2010, 2009–2016 CrossRef.
- R. H. Perry, M. Splendore, A. Chien, N. K. Davis and R. N. Zare, Angew. Chem., Int. Ed., 2010 DOI:10.1002/anie.201004861.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary information. See DOI: 10.1039/c0sc00416b |
| This journal is © The Royal Society of Chemistry 2011 |