The unique nature of H+ in water†
Evgenii S.
Stoyanov
*,
Irina V.
Stoyanova
and
Christopher A.
Reed
*
Department of Chemistry, University of California, Riverside, CA 92521, USA. E-mail: evgeniis@ucr.edu; chris.reed@ucr.edu
First published on 16th December 2010
Abstract
The H+(aq) ion in ionized strong aqueous acids is an unexpectedly unique H13O6+ entity, unlike those in gas phase H+(H2O)n clusters or typical crystalline acid hydrates. IR spectroscopy indicates that the core structure has neither C3vH3O+ Eigen-like nor typical H5O2+ Zundel-like character. Rather, extensive delocalization of the positive charge leads to a H13O6+ ion having an unexpectedly long central O⋯O separation of ∼2.57 Å and four conjugated O⋯O separations of ∼2.7 Å. These dimensions are in conflict with the shorter O⋯O separations found in structures calculated by theory. Ultrafast dynamic properties of the five H atoms involved in these H-bonds lead to a substantial collapse of normal IR vibrations and the appearance of a continuous broad absorption (cba) across the entire IR spectrum. This cba is distinguishable from the broad IR bands associated with typical low-barrier H-bonds. The solvation shell outside of the H13O6+ ion defines the boundary of positive charge delocalization. At low acid concentrations, the H13O6+ ion is a constituent part of an ion pair that has contact with the first hydration shell of the conjugate base anion. At higher concentrations, or with weaker acids, one or two H2O molecules of H13O6+ cation are shared with the hydration shell of the anion. Even the strongest acids show evidence of ion pairing.
Introduction
The state of H+ in water (frequently referred to as the “proton” or “excess proton” but more properly called the hydrogen ion) is one of the oldest unsolved problems in chemistry.1 The earliest studies centered on the unusually fast mobility of H+ in galvanic cells. In a remarkable tribute to the importance of ideas, the name of Grotthuss is associated with the proton hopping mechanism that grew out of his description of charge diffusion over 200 years ago,2 at about the same time that Gay-Lussac confirmed that water was H2O not HO!3 Mobility studies were used by Eigen4 in the mid 20th century to formulate H+(aq) as a C3v trihydrated H3O+ ion and this formulation persists in textbooks today despite the rather restrictive conditions now known for its existence.5–7 With the availability of IR spectrometers in the 1950s, attempts were made to measure and interpret the IR spectrum of H+(aq).8,9 These met with mixed success because it was not possible to cleanly isolate the spectrum of the H+(aq) cation from that of water solvent and the evident broad absorptions in these qualitative spectra defied easy interpretation. Zundel put forward compelling arguments for the existence of an alternate structure for H+(aq) based on the short, strong, symmetrical H-bonding in the H5O2+ ion which now bears his name.8 Despite extensive experimental effort in condensed phases into the 1970s, the true nature of H+(aq) in water remained inconclusive. A rapid equilibrium between Eigen- and Zundel-type ions became a popular hypothesis:10–14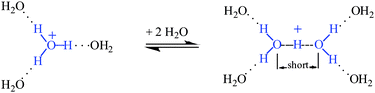
Calculated ratios of Eigen versusZundel ions of ca. 65![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3515,16 or 75:2514 have been suggested. The isolation of crystalline forms of each of these ions in symmetrical anion environments lent credence to their proposed existence in the liquid phase and X-ray structure determinations defined their O⋯O separations: ∼2.55 Å in Eigen-type ions17,18 and ∼2.39 Å and ∼2.52 Å in Zundel-type H5O2+·4H2O ion for the central and peripheral O⋯O separations, respectively.19
:3515,16 or 75:2514 have been suggested. The isolation of crystalline forms of each of these ions in symmetrical anion environments lent credence to their proposed existence in the liquid phase and X-ray structure determinations defined their O⋯O separations: ∼2.55 Å in Eigen-type ions17,18 and ∼2.39 Å and ∼2.52 Å in Zundel-type H5O2+·4H2O ion for the central and peripheral O⋯O separations, respectively.19
Meanwhile, theoretical and gas phase experimental methods for studying mass-isolated H+(H2O)n clusters were advancing rapidly and these approaches have dominated research on the hydrated proton for the past couple of decades.16,20–28 Elegant experimental studies have been combined with theory to establish the gas phase structures and IR spectra of H+(H2O)n clusters up to n = 100 values16,20–26,28 with the expectation that these results provide information relevant to H+ in liquid water. The most recent theory29,30 for large clusters supports the predominant existence of a hybrid structure that can be described as an Eigen ion distorted towards a Zundel-type ion by having one short O⋯O separation (the “special pair”). The short O⋯O bond fluctuates over all three Eigen-type O⋯O bonds on the picosecond timescale so IR spectroscopy, which has >100 ps timescale, is not expected to detect this asymmetry in normal mode vibrations.
We now bring these studies full circle by applying quantitative IR techniques to H+(H2O)n in liquid water. We come to the surprising conclusion that the accumulated decades of circumstantial evidence for the structure of H+(H2O)n in liquid water are inconsistent with our new IR and X-ray data. These data indicate that delocalization of positive charge in liquid water is more extensive than could have anticipated from gas or solid phase studies or by theory. This is evidenced by O⋯O bond elongation such that neither traditional Eigen nor Zundel structures are consistent with the data. Ion pairing is also more prevalent than previously believed.
The present work addresses the following questions.
(1) What is the value of n in H+(H2O)n that adequately describes H+(aq)? Surprisingly, this question has not been addressed. It is synonymous with an enquiry into the extent of positive charge distribution over the hydrations shells of the H+(aq) ion. We use the criterion that if the IR spectrum of a water molecule is different from that of bulk water, it is part of the solvation shell of an ion.
(2) What is the true IR spectrum of H+(aq)? What are the correct vibrational assignments of the bands and what is the origin of the mysterious continuous broad absorption (cba)? Theories developed during the 1970–80s to explain the cba are many. They include Zundel's theory of strong OH+O bond polarisability whereby the proton interacts with the fluctuating electric field of the surroundings,8,31 stochastic theory,32,33 combined transitions of the proton in H5O2+ accompanied by multiple excitation of low frequency oscillators of the hydration shell,34,35 strong interaction between the ν(O–H) and ν(O⋯H) vibrations,36 anomalous amplification of a large number of overtones and combinations,37,38 and a rapid interconversion between H3O+ and H5O2+ within a special OH+O bond.39 In the present paper, we redefine the cba and its origin based on new experimental data.
(3) What is the molecular structure of H+(aq) giving rise to its IR spectrum? At a minimum, the locations of the oxygen atoms need to be determined and compared to Eigen- and Zundel-type ions.
(4) What is the role of ion pairing on the structure of ionized strong acids? We note that gas phase studies and, with few exceptions,40,41 theoretical calculations are carried out in the absence of anions. We have recently shown that in chlorocarbon solvents, where tight ion pairing is important, neither Eigen nor Zundel-type core ions exist for H(H2O)n+ (n = 3–8) clusters. Rather, the core ion is the H7O3+ cation.42
These questions are addressed by IR spectroscopy using quantitative methods where only qualitative had been applied previously.43,44 A method for determining the value of n in H+(H2O)n was developed some time ago to determine the stoichiometry of H+(aq) incorporated into the water core of reverse micelles.45,46 More recently, we communicated the results of applying this method to dilute aqueous solutions of carborane superacids and HClO4 and found n = 6.47 In the present work we extend these studies to common mineral acids such as HCl, HNO3 and to higher concentrations of HClO4. Also in the recent communication, we published the first quantitatively reliable IR spectrum of H+(aq) cation and offered a preliminary interpretation in terms of a unique H13O6+ structural entity having O⋯O separations that are longer than those in either Eigen or Zundel ions, and longer than those predicted by theory. In the present paper, we offer a more complete analysis of the IR spectrum including a new explanation for the continuous broad absorption (cba). Finally, we relate these IR studies to the unique X-ray structure of the carborane acid H(CHB11I11)·8H2O48 whose nanotubular structure appears to offer the first good structural model for H+(aq). Unlike all other X-ray structures of H+(H2O)n+ salts, it has O⋯O separations that reflect the delocalized, mobile nature of H+ in liquid water. They are significantly longer than those found the more localized, static structures having traditional Eigen or Zundel character.
We use attenuated total reflectance (ATR) infrared spectroscopy because in transmission mode the spectrum of water has a very high molar absorptivity in the OH stretching vibration region, resulting in saturation. ATR is regarded as the best method for high-precision measurements in liquid water.49,50 The depth of IR penetration is a few microns. Effects of the diamond surface on dissolved electrolytes are confined to a few nanometers so they are not registered by ATR IR.
Experimental
HClO4, HNO3 and HCl acid were used without additional purification. Acid concentrations were determined by titration with standardized NaOH solutions.IR spectra were recorded in ATR mode by placing a drop of solution onto a diamond crystal using a Perkin Elmer Spectrum-100 spectrometer in the 4000–400 cm−1 frequency range. Raw spectra were corrected using the ATR algorithm51 and manipulated using GRAMMS software.
Results
Isolating the IR spectrum of H+(H2O)n
The IR spectrum of a strong aqueous acid consists of the spectrum of the anion and overlapping spectra from three types of water molecules: bulk water, water associated with Haq+, and water perturbed by the anion. The spectrum of the anion and water perturbed by the anion can be readily subtracted using the spectrum of the corresponding alkali metal salt. Because the spectrum of the hydration shell of an alkali metal cation happens to coincide with that of bulk water52 it is successfully subtracted along with the solvent. In this manner, the spectra of the hydration shells of anions CHB11Cl11−, ClO4−, NO3− and Cl− (Fig. 1) have been isolated for aqueous solutions of their sodium and caesium salts. When normalized to unit concentration these spectra coincide, indicating that they are independent of the nature of cation (Na+ or Cs+) in the concentration range studied. This is illustrated for Cl− in Fig. 2.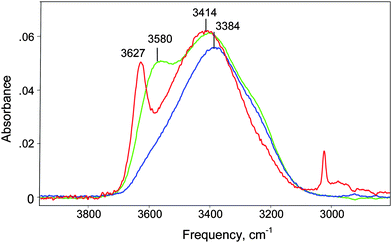 | ||
| Fig. 1 IR spectra of the first hydration shells of anions Cl− (blue), ClO4− (green) and CHB11Cl11− (red) normalized to unit concentration. These were obtained by subtracting the spectrum of bulk water from the spectra of water solutions of their sodium salts (1.5 M for Cl−, ClO4− and 0.446 M for CHB11Cl11−). | ||
 | ||
| Fig. 2 IR spectra of the hydration shell of Cl− obtained after subtracting the spectrum of bulk water from those of aqueous solutions of NaCl (1.0 and 2.0 M) and CsCl (0.5, 1.0 and 2.0 M). Spectra are normalized to unit concentration. | ||
Since aquated anions, Aaq−, have essentially no influence on the H-bonded network of liquid water beyond the primary hydration shell53,54 the spectra in Fig. 1 belong to their first hydration shell and are easily interpreted. The highest frequency νOH band belongs to the O–Hagroup of water molecules directly H-bonded to anion:
| A−⋯Ha–O–Hb⋯(OH2) |
The O–Hb group is H-bonded with outer sphere water molecules and its frequency is practically indistinguishable from that of bulk water.53
The νOHa frequency reflects the proton acceptor properties of an anion and, as shown in Fig. 3, correlates very well with the ΔνNH basicity scale for anions based on trioctylammonium salt ion pairs.55 For Claq−, the νOHa frequency is the lowest compared to all other anions studied and strongly overlaps with νOHb, giving rise to a single band at 3384 cm−1. This confirms that Cl− is the most basic anion of those studied and its conjugate acid in water is correspondingly the weakest.
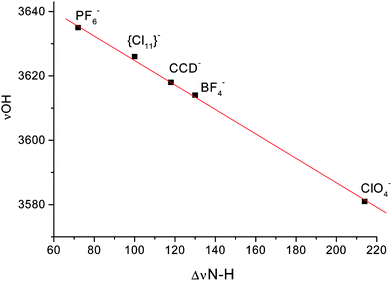 | ||
| Fig. 3 The νOHa frequency dependence of water bounded to anion on the ΔνNH scale basicity of anions55 | ||
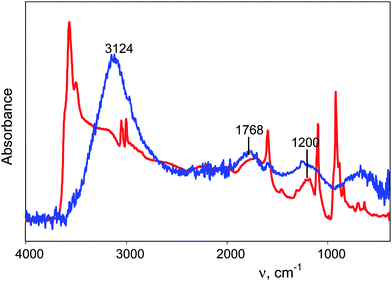
These considerations allow the isolation of well-grounded spectra of H+(H2O)n cations. There are two steps. First, from the spectrum of the acid HA solution (as illustrated for HNO3 in Fig. 4, black), the spectrum of equimolar NaA is subtracted with a scaling factor f = 1. This removes the spectra of bulk water, the anion and water associated with the anion. The resulting spectrum belongs to H+(H2O)n but is distorted (Fig. 4, blue) because the spectrum of bulk water is over-subtracted by an amount equal to the number of water molecules involved in the H(H2O)n+ cation. Thus, in the second step, the spectrum of the bulk water is added with increasing scaling factor up to full removal of the spectral distortion. The criterion for full removal of the spectral distortion is strict compensation of the δH2O band at 1632 cm−1 using the method45,46 of equality of areas −S and +S (Fig. 4 inset, barred regions in red spectrum). The resulting spectrum (Fig. 4, red) is the true spectrum of the H+(H2O)n cation, blemished only by residual inflection point distortions from the −S and +S subtraction process which reflect otherwise imperceptible changes in the IR spectrum of water molecules not included in the H+(H2O)n formulation. Deconvolution of the IR spectrum (Fig. 5 red) gives the continuous broad absorption (cba, blue) overlaid with Gaussian bands (green) with frequency variances for all acids studied in dilute solution (≤0.5 M) of 3120 ± 20, 2816 ± 40, 1747 ± 5, 1202 ± 5 and 654 ± 12 cm−1.
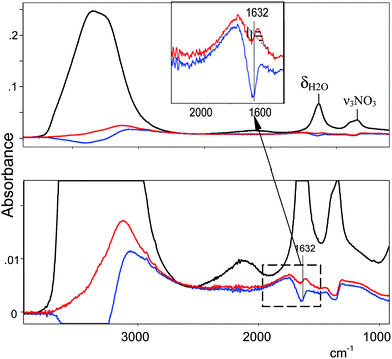 | ||
| Fig. 4 The procedure for quantitative isolation of the IR spectrum of H+(H2O)n cations illustrated for HNO3. From the spectrum of 0.75 M HNO3 (black) was subtracted the spectrum of 0.75 M NaNO3 resulting in spectrum (blue). Addition of the spectrum of water to (blue) up to equality of the intensities of −S and +S for the δH2O band at 1632 cm−1 (inset) leads to spectrum (red), the true spectrum of the H+(H2O)n cation. | ||
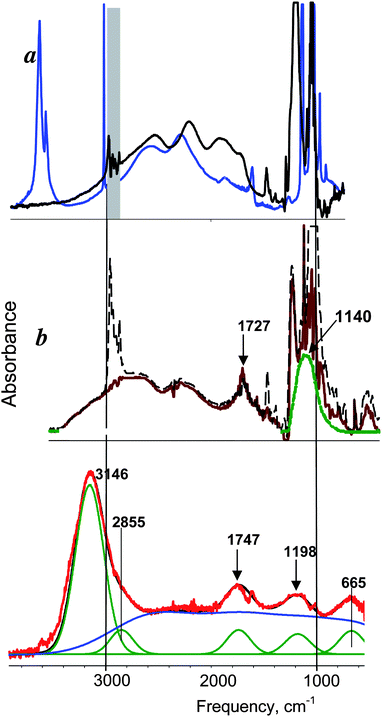 | ||
| Fig. 5 IR spectrum of H+(H2O)6 cation in water (red) compared with those of: a) H3O+·3H2O in crystal H+(H2O)4{Cl11} (blue) and (H3O+·3TBP)FeCl4− in CCl4 solution (black); b) H5O2+·4TBP in TBP solutions of H5O2+ClO4− salt, initial (black dashed) and after subtraction of C4H12O groups absorption of TBP. | ||
Determination of n in H+(H2O)n
The isolation of the spectra of H+(H2O)n cations allows simultaneous determination of the stoichiometry n. As previously described in Supp. Info.,47† the concentration of water molecules incorporated into the H+(H2O)n cation can be determined from equation:| CH2Obound = CH2OHA − CH2ONaA + 55.455 × f | (1) |
As communicated earlier,47 the value of n in the H+(H2O)n cation was found to be equal to 6 for the strongest acids. This includes all carborane acids as well as HClO4 at concentrations below 0.7 M. The n values are given in Table 1 for HClO4 at higher concentrations and for HNO3 and HCl acids. They show decreasing n values with increasing concentration. As discussed later, these are ascribed to ion paring effects in these weaker acids.
| Concentration, mol L−1 | Acid | ||
|---|---|---|---|
| HClO4 | HNO3 | HCl | |
| 0.5 | 5.98 | 6.33 | 5.1 |
| 0.7 | — | — | 5.1 |
| 0.75 | 5.88 | 5.73 | — |
| 1.0 | 5.84 | 6.03 | 5.02 |
| 1.5 | 5.78 | 5.31 | 5.1 |
| 2.0 | 5.67 | 4.89 | 5.22 |
| 3.0 | 5.45 | 4.54 | 5.18 |
| 4.0 | 5.43 | 4.35 | 5.5 |
Discussion
The structure of H+(H2O)n and its IR spectrum
The determined stoichiometry of the H+(H2O)6 cation in strong acids along with a preliminary interpretation of its IR spectrum led us to formulate Haq+ as a unique H13O6+ ion having the structure shown schematically in 2D in Scheme 1 (blue).47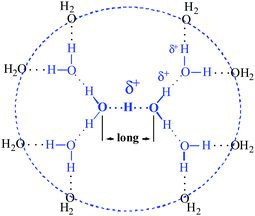 | ||
| Scheme 1 Schematic 2D structure of the H13O6+ ion. | ||
Let us now discuss this in more detail with additional evidence. Obviously, the n = 6 result rules out formation of a C3v Eigen-type H3O+·3H2O ion because it has a stoichiometry of n = 4. Moreover, the IR spectrum of the H+(H2O)6 cation is quite different from that of Eigen-type ions. These include H3O+ bound to three water molecules in crystallographically characterized [H3O+·3H2O][CHB11Cl11−]42 and H3O+ bound to three tributylphosphate molecules (TBP) in [H3O+·3TBP][FeCl4−].57
The basicity of TBP is known to be very close to that of bulk water57 so the IR spectra of these ions are expected to be very similar. As seen in Fig. 5a (blue, black), the central H3O+ group of these Eigen-type ions has a characteristic broad absorption in the 1700–3000 cm−1 frequency range42 which is absent in the spectrum of the H+(H2O)6 cation (Fig. 5, red). Clearly, C3v Eigen-type H3O+ ions do not exist in aqueous acid solutions.
Since the n = 6 stoichiometry of the H+(H2O)6 ion is compatible with a tetrahydrated Zundel-type H5O2+ · 4H2O ion, the appropriateness of a Zundel-type formulation for Haq+ must be made on the basis of spectral similarity. This is somewhat problematic because, while structurally characterized by X-ray crystallography,19 the IR spectrum of the Zundel-type H5O2+ · 4H2O ion has not been reported in condensed phases. Nevertheless, the IR spectrum of the H5O2+ ion surrounded by four TBP molecules is known57 and the close similarity of the basicity of TBP with that of bulk water leads to the expectation that the spectrum of H5O2+·4TBP will be a good model for that of H5O2+·4H2O. The comparison is shown in Fig. 5 (red vs. brown). There is a similarity, but also an important distinction. The similarity is the development of the two Zundel ion marker bands, in the ranges 1140–1200 and 1727–1747 cm−1, which are associated with the vibrations of the central symmetric O–H+–O group with strong H-bonds.58 However, these frequencies for the H+(H2O)6 cation are significantly higher than those of the H5O2+·4TBP cation.
It is known that the frequencies of both Zundel ion marker bands increase linearly with increasing the basicity of L-ligands of H5O2+·4L because of decreasing positive charge and decreasing H-bond strength at the central OHO group.58 As shown in Fig. 6, the lower frequency band, νasOHO, increases from ∼1060 cm−1 in the bare (gas phase) H5O2+ ion59,60 to 1140 cm−1 for L = tributylphosphate, whose basicity coincides with that of liquid water.57 It increases further to 1160 cm−1 for L = phosphine oxides, whose basicities exceed that of water.61 This represents the limit of existence of H5O2+·4L because with this L basicity there is an equilibrium with proton disolvate-type L–H+–L ion.61 If the basicity of L exceeds that of phosphine oxides, then only L–H+–L ions exist. The νasOHO value for H+(H2O)6 in water lies conspicuously off the basicity correlation in Fig. 6 (red asterisk). Its high frequency of 1198–1200 cm−1 corresponds to a region where typical Zundel-type H5O2+ ions do not exist. The frequency indicates that the central O–H–O group has both anomalously low positive charge and an anomalously long O⋯O distance compared to typical Zundel-type ions.
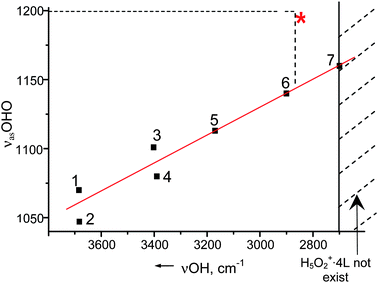 | ||
| Fig. 6 The νasOHO dependence on basicity of L in H5O2+ · 4L cations. 1, 2—bare in gas phase; 3—{Cl11} ion in solid state; 4—C6H6 in benzene solution; 5—diethyl ether in benzene solution; 6—tributyl phosphate in CCl4 solution; 7—phosphinoxide in dichloroethane solution. | ||
The possibility of such structural peculiarity in the (H13O6+)aq cation was recently discovered in the X-ray structure of H(CHB11I11)·8H2O, where three unusual types of protonated water clusters are formed in water filled nanotubes (Fig. 6b).48 These are the (H13O6+)aq ion (I), the H7O3+·H2O ion (II) similar to that formed by ion-paired hydrated carborane acids in organic solvents42 and the previously unknown square-planar (H17O8+)aq cluster (III). All clusters have unprecedented, long O⋯O distances. The IR spectrum of this crystal, which represents the overlapping spectra of these three cluster types, shows the same band at ca. 1200 cm−1 seen in the spectrum of H13O6+ in an aqueous solution of H(CHB11I11) (Fig. 7). Does this band belong to only cluster I or do clusters II and III also contribute to this absorption? In Fig. 8 the spectrum of crystalline H(CHB11I11)·8H2O is compared to that of the H7O3+·5H2O cation formed in water-saturated dichloroethane solution of H(CHB11I11).42 One can see that the H7O3+·5H2O cation, which is topologically analogous to cluster II and has an O⋯O distance of ∼2.5 Å,42 has no absorbance in the ∼1200 cm−1 region. Since both clusters II and III have longer O⋯O distances of ∼2.62 and ∼2.64 Å, respectively, they too will not have absorptions in this region. Thus, we conclude that the ∼1200 cm−1 band in the spectrum of crystalline H(CHB11I11)·8H2O arises from the νasOHO frequency of the central OHO group of cluster I. Since H13O6+ ions in both this crystal and in liquid water are surrounded by water molecules and their νasOHO absorption have coincident frequency and similar width/shape, they must have a similar degree of positive charge delocalization and a similar central O⋯O and four conjugated O⋯O distances. This allows us to define the structure of the H13O6+(aq) ion in Scheme 1 more exactly. Its key features are an unprecedented long central O⋯O distance of ∼2.57 Å and O⋯O distances of four conjugated H-bonds with ∼2.7 Å.
 | ||
| Fig. 7 IR spectrum of H13O6+ in water solution of H(CHB11I11) acid (the absorption of anion is subtracted) (blue) in comparison with spectrum of crystal H(CHB11I11)·8H2O (red). | ||
 acid in dichloromethane solution (blue).](/image/article/2011/SC/c0sc00415d/c0sc00415d-f8.gif) | ||
| Fig. 8 IR spectrum of crystal H(CHB11I11)·8H2O (red) in comparison with spectrum of [H7O3+ · 5H2O](CHB11I11) acid in dichloromethane solution (blue). | ||
The other characteristic IR bands of the (H13O6+)aq cation are readily interpreted by analogy to H5O2+ · 4L cations: the 1768 cm−1 band, like that at ∼1200 cm−1, is associated with the central OHO group, the 2855 cm−1 band to the OH stretch of the two H2O molecules forming the central H5O2 group and the 3146 cm−1 band is assigned to the OH stretch of the four peripheral H2O molecules.
H/D comparison
A comparison of the band frequencies of protio and deutero forms of the H13O6+(aq) cation (Table 2) shows that the H/D isotopic ratios for both OH stretching and torsional vibrations are close to 1.35, i.e. consistent with harmonic vibrations. However, the two bands arising from the central OHO group vibrations have lower H/D ratios indicating that they are anharmonic. This provides an additional criterion on which to judge the appropriateness of theoretical determinations of structure. We note that isotopic H/D ratios bands of the trihydrated Eigen ion (see Supp. Info.†) are all in the range common for harmonic vibrations (1.34–1.35).| Vibrations | X = H | X = D | H/D |
|---|---|---|---|
| νOX peripheral | 3146 | 2340 | 1.35 |
| νOX of H5O2 group | 2855 | 2111 | 1.35 |
| δOXO | 1747 | 1406 | 1.24 |
| ν asOXO | 1198 | 922 | 1.30 |
| Torsion | 672 | 491 | 1.36 |
Ion pairing effects
Infrared spectra of aqueous solutions of the studied acids as a function of concentration in the 0.2–4 M range show good isosbestic points. These occur at 3146 cm−1 for HCl, 3542 cm−1 for HNO3 and at 3518 and 3072 cm−1 for HClO4 (see Supp. Info.†). This might be taken as evidence that only one type of H+(H2O)n cluster is present in these acids. However, isosbestic points are relatively insensitive to small changes in the intensities of bands. By careful attention to the quantitative subtraction of the spectrum of bulk water we find that n values decrease with increasing concentration for two acids, HNO3 and HClO4. This is ascribed to ion pairing effects.Increasing the HClO4 and HNO3 acid concentrations up to 4 M leads to gradual decreases in n from 6 to 5.4 and 4.3, respectively (Table 1). This indication of stronger ion pairing effects in HNO3 relative to HClO4 is consistent with the higher basicity of NO3− on the ΔνNH scale55 and with the lower acid strength of HNO3 (pKa -1.5) relative to HClO4 (pKa -8). In their IR spectra, normalized to unit concentration, the change in n value is reflected mainly in a proportionate decrease in the intensity of the νOH absorption at 3120–3180 cm−1, the band associated with the four peripheral H2O molecules of the H13O6+ ion. The remaining part of the Haq+spectrum is insignificantly changed. This is illustrated for HNO3 in Fig. 9. For HCl, the constant value of n = 5 and the invariability of the normalized IR spectra over the 1–4 M concentration range indicates constant ion pairing (Supp. Info†).
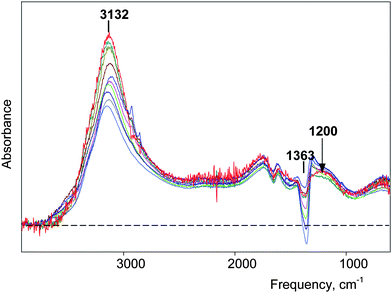 | ||
| Fig. 9 IR spectra of H(H2O)n+ clusters normalized to unit concentration for HNO3 water solutions with concentrations varied from 0.25 (red) to 4.0 M (blue). The inaccurate subtraction of ν3NO3 band (over subtraction at 1363 cm−1) arises because of different concentration dependence of the spectrum of NO3− anion for studied HNO3 solutions and used for subtraction NaNO3 solutions. | ||
Taken all together, these data suggest that ion pairing perturbs up to two of the four peripheral water molecules of the H13O6+ ion, removing their IR spectral peculiarity i.e., restoring the distinctively absent δH2O band. As discussed in more detail later, absent IR bands are associated with ultrafast proton motion. Shared water molecules in an ion pair (Scheme 2) between H(H2O)n+ and the first hydration shell of the anion, where water mobility is slow,53,54,62 will immobilize these water molecules and restore their δH2O band. Thus, the spectrum of the shared water molecules is subtracted as the hydration shell of anion and the determined stoichiometry of H+(H2O)n has n < 6.
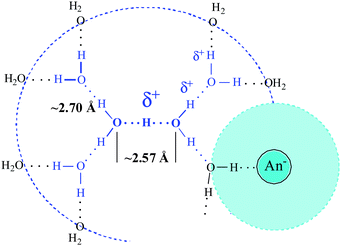 | ||
| Scheme 2 Ion pairing with shared water molecule. | ||
The basicity of the O-atoms of the shared water molecules should increase slightly compared to that of the remaining unshared peripheral water molecules of the H+(H2O)n cation because, as indicated by Fig. 1, H-bonding with Cl−, NO3− or ClO4− anion is weaker than with outer sphere water molecules. However, this change in O-basicity is apparently too weak to notably influence the spectrum of H+(H2O)n cation when passing from n = 6 to 5 or 4, other than decreasing the intensity of the νOH band at ∼3100–3200 cm−1 (Fig. 9). This indicates that the n = 4 cation retains high spectral and structural similarity with n = 5 and 6.
While the δH2O bands of the shared and peripheral water molecules are insensitive to their nonequivalence, the OH stretches are much more sensitive. The νOH bands of the shared water molecules should lie between that of the unshared peripheral H2O molecules (3100–3200 cm−1) and νOH of unshared water molecules bound to the anion. Thus, these bands will not be perfectly subtracted. This is detected as a weak distortion on the high frequency side of the νOH absorption at ∼3100–3200 cm−1 (Fig. 9). As expected, the distortion increases with decreasing n in H+(H2O)n.
Corroborating evidence for ion pair formation is found in the IR spectrum of anion, most sensitively with the NO3− anion. The decrease in its symmetry under the influence of a unidirectional electrostatic interaction splits the doubly degenerate ν3NO3 stretching band. The stronger this interaction, the larger is the Δν3 splitting. As shown in Fig. 10, Δν3 increases with increasing HNO3 concentration starting at ca. 0.6 M. This is consistent with decreasing average stoichiometry in H+(H2O)6 below 6. At HNO3 concentrations below 0.6 M, where only the n = 6 cation is formed, Δν3 appears to be concentration independent. Nevertheless, the residual Δν3 splitting (∼54 cm−1) indicates that ion pairing must still be present. A model that accommodates these data is shown in Scheme 3. The H13O6+ cation and inner solvation shells of the anion remain intact but electrostatic contact is sufficient to perturb the D3h symmetry of NO3− anion.
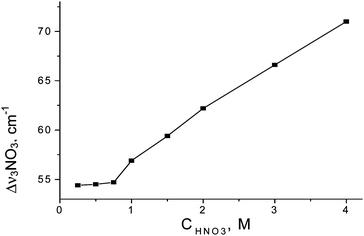 | ||
| Fig. 10 Dependences of Δν3NO3 splitting on HNO3 concentration. | ||
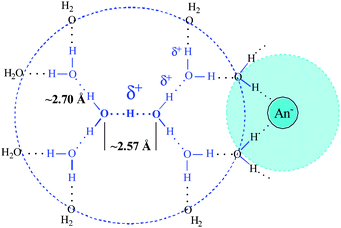 | ||
| Scheme 3 Ion pairing in dilute solution. | ||
The triple degenerate ν3 stretch of the ClO4− anion changes with increasing perchloric acid concentration in a similar manner to the ν3NO3 band although the splitting is less pronounced. This indicates the same type of ion pairing is occurring.
Carborane superacids are the strongest known acids63 so ion pairing is expected to be the weakest or non-existent. Indeed, the IR spectrum of the carborane anion is not detectably dependent on acid concentration and only H13O6+ cations are formed. Nevertheless, the νOH frequency of the four peripheral H2O molecules of the H13O6+ cation shows a small dependence on the nature of carborane anion47 that exceeds the error of νOH determination. Thus, even ionized carborane acids, (H13O6+)Carb−aq, appear to be ion paired in the manner of Scheme 3. It is likely that all acids, no matter how strong, have ion pairing of this type.
For dilute solutions of mineral acids (< 0.7 M), the intensity of νOH bands (IOH) can be measured more precisely than the determination of n in H+(H2O)n. That is why the dependence of IOH on CHA can give more precise information about composition of Haq+ at low concentrations, where stoichiometry cannot be accurately determined, and where n starts to change from 6 to 5. The IOH dependences on acid concentration (CHA) for HCl, HNO3 and HClO4 are given in Fig. 11. They show more sharply that ion pairing of the type in Scheme 2 with n = 5 begins at ∼0.5 for HNO3 and ∼0.9 M (HClO4). Below these concentrations, ion pairing only occurs with the n = 6 cation according to Scheme 3. In the case of HCl, from as low as 0.3 M and up to 4 M concentration, only ion pairing with n = 5 is observed (Scheme 2). Thus, in spite of the leveling of acid strength in dilute solutions of these strong acids, measurable differences in ion paring formation with n = 5 or 6 nevertheless reflects their relative acid strengths: H(carborane) > HClO4 > HNO3 > HCl.
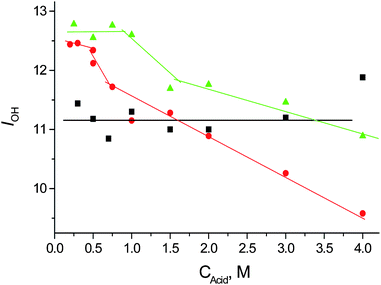 | ||
| Fig. 11 Dependences of OH stretch intensities (IOH) on acids concentration. Green—HClO4; red—HNO3; black—HCl. | ||
The uniqueness of H+(H2O)6 cation
This is best revealed by comparison to the protonated methanol cluster, H+(CH3OH)8, formed when a strong acid ionizes in neat methanol.64 The 3D-structured H+(H2O)6 cation has important similarities, as well as important distinctions, from 1D-structured H+(CH3OH)8 cation shown in Scheme 4.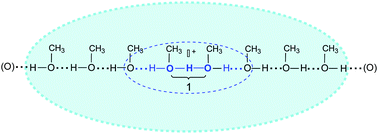 | ||
| Scheme 4 Representation of H(CH3OH)8+ cation structure in neat methanol. | ||
The similarities in their IR spectra (Fig. 12) are: (i) both cations have characteristic bands at 1567–1747 and 958–1197 cm−1 arising from the central OHO group vibrations and νOH stretch frequencies of all other OH groups in the 2500–3200 cm−1 region, and (ii) bands from δCOH bending and νCO stretching vibrations of H+(CH3OH)8 are absent in its IR spectrum just as the δH2O bands are absent in the spectrum of H(H2O)6+. These similarities are common to all proton disolvates L–H+–L, including H5O2+·4L (L = nonprotic base) and have been discussed earlier.58,64–66 This peculiar absence of bands defines the sharp boundary between the eight CH3OH molecules in H+(CH3OH)8 or the six H2O molecules in H13O6+ and the surrounding molecules of bulk methanol or water, respectively. Absent CO stretch and COH bend vibrations bands in H+(CH3OH)n cations are characteristic of chain-linked methanol molecules conjugated with the central O–H+–O group.64 Even the CH stretching vibrations of the methyl groups are affected. Compared to the intensity of common CH3OH molecules, their intensity decreases by 94% for those associated with the central O–H+–O group and 60% for those associated with the conjugated O–H⋯O groups. To date, this has not been addressed by theory but must be connected with special dynamic properties of the central OHO group.
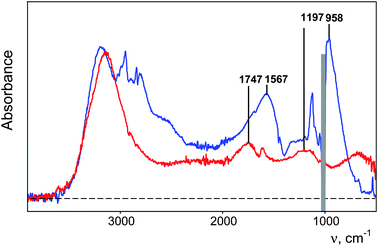 | ||
| Fig. 12 IR spectra of 0.4 M H(CHB11Cl11) solutions in water (red, anion absorption is subtracted) and methanol (blue). | ||
Regarding the important distinctions between 3D H(H2O)6+ and 1D H+(CH3OH)8 clusters, these are mainly concerned with the central O–H–O group and its response to conjugated peripheral molecules. In the case of H+(CH3OH)n clusters when passing from n = 2 to 4, νasOHO increases from 950 to 1010 cm−1 without changing in intensity (Table 3). This occurs because of partial delocalization of the positive charge onto the two new O–H⋯O groups, accompanied by decreasing charge on the central OHO group and increasing O⋯O distance. For the next increases of n up to 8, there is practically no more influence on the central O–H+–O group (νas and IOHO are constant). However, replacement of benzene solvent with methanol for the cluster with n = 8 returns νas to the initial value of 950 cm−1 as seen in the n = 2 cluster, i.e., the positive charge and O⋯O distance are restored. In the case of H(H2O)n+ clusters the situation is the opposite. Increasing n from 2 in benzene to 6 in water results in a high frequency shift of ΔνasOHO by 170 cm−1 whereas there is no shift for the corresponding H+(CH3OH)n clusters with n = 2 in benzene and n = 8 in methanol (Table 4). It is clear that the 3D water cluster structure favors positive charge delocalization from the central OHO group much more than the 1D methanol structure. The difference lies in their outer sphere solvation and ion pairing. The 3D water cluster has eight H-bonded outer sphere water molecules onto whose O-atoms the positive charge can be delocalized whereas methanol clusters are restricted to charge delocalization in 1D and rely on greater accessibility of the central O–H+–O group to ion pairing and electrostatic solvation with outer sphere methanol.64 This makes the H13O6+aq ion unique and causes an unprecedented increase in the O⋯O distance of the central OHO group.
Another important distinction follows from the intensities of the νasOHO band (IOHO). In the case of H+(CH3OH)n clusters, increasing n from 2 to 8 does not change IOHO significantly (Table 3). The same is true when passing from H5O2+·4C6H6 in benzene solution to H(H2O)6+ in solid state (Table 4). However, for H(H2O)6+ in aqueous solution, IOHO is more than a factor of three lower (Table 4) and it is accompanied by the appearance of the continuous broad absorption (cba). In other words, only about 30% of absorption of H(H2O)6+aq is observed as absorption bands due to normal vibrations, the other 70% appears to be developed as a continuum. In contrast, the H+(CH3OH)8 cation in neat methanol develops normal IOHO intensity for O–H+–O group vibrations and does not develop a cba, only a broad absorption from multiple OH vibrations that is often mistakenly named a cba.
The continuous broad absorption (cba)
These observations allow us to give definition to the cba and ascertain its nature. The cba is an unstructured continuous absorption that develops in IR spectra of acidic water solutions across the entire 3000–50 cm−1 frequency range (Fig. 5, blue). Attempts to divide it into separate broad bands67 are unconvincing. Such cba is not observed in the IR spectra of H3O+·3L, H5O2+·3L and H7O3+·5L cations with L = organic basics or H2O, in the solid state or in solutions of organic solvents.5,42,48,58 When the basicity of L in these species is high (H2O, tributylphosphate, phosphine oxides), their stretching OH vibrations develop as very broad overlapping bands in the 3000–1200 cm−1 frequency range having the so-called ABC structure68 which has often been mistaken for cba. This is common to all species with asymmetric O–H⋯O groups having O⋯O distance ca. 2.5–2.6 Å.42 These broad absorptions are quite distinct from the cba of acidic water solutions by not being continuous and featureless. They can be deconvoluted into separate bands. To the best of our knowledge, cba is a specific spectroscopic feature found only in aqueous H(H2O)n+ clusters in liquid water. The spectrum of the H(H2O)6+ cluster in the solid state has no cba and the intensity of the absorption bands from normal vibrations is undiminished (Table 4). Similarly, such intensity conversion from cba to normal mode vibrations has been detected by Raman spectroscopy when 4M aqueous HCl or HBr form glasses at liquid nitrogen temperature. The cba intensity significantly decreases with temperature and is accompanied by a simultaneous increase in the intensity of the band at ∼1200 cm−1.69 These observations indicate that the cba is associated with the dynamic properties of excess proton. At low temperature in a glacial state (−180 °C), or in the crystalline state at room temperature, the translational movements of the protons in the H(H2O)6+ cation are suppressed and the cluster develops normal vibrations. On the other hand, at room temperature in liquid water, proton dynamics are comparable to the timescale of vibrational transitions.13 As a consequence, the rates of vibrational transitions involving the five internal H-atoms of H(H2O)6+ exceed the lifetimes of their fixed positions. On the IR timescale, the location of these five H-atoms becomes uncertain and a continuous absorption develops instead of line spectra from normal vibrations. Since the intensity of the absorption bands of Haq+ in water corresponds to ∼30% of that for normal vibrations (Table 4), on average only in ∼30% of H13O6+ clusters do the 5 H-atoms have lifetimes of their fixed positions sufficient to produce normal vibrations. The other ∼70% of H13O6+ clusters develop a cba. Shifting the equilibrium percentages between the line spectrum and the cba requires a significant temperature change that exceeds the liquid range of water. That is why small increases in temperature of water solutions from 293 to 313 K67 or 363 K70 do not reveal changes in the cba.Dynamic properties of the excess proton may also be responsible for the disappearance of the δH2O band at ∼1630 cm−1 for all 6 H2O molecules in the H13O6+ cation, as well as the νCO and δCOH bands of H+(CH3OH)n clusters and related bands in proton disolvates H+L2.58,61,64–66 This effect is observed only in species having a symmetrical O–H+–O group with a flat-bottomed proton potential function. It was initially discovered in crystalline di(methylphenyl)phosphoric or di(chlorophenyl)phosphoric acid dimers, where the both νasPOO and νsPOO bands disappear in the IR spectra and the equivalency of the O-atoms in the O–H+–O group was established by X-ray crystallography.71–73 The phenomenon is not dependent on O⋯O distance since they differ significantly in these two acid dimers, 2.512 Å72 and 2.398 Å respectively.74 In contrast, strong bands from νPOO are observed in the IR spectra of dimeric dialkylphosphorous or dialkylphosphinic acids where the OHO groups are unsymmetrical.71,72 Similarly, the δHOH frequencies disappear in symmetric H5O2+58 but not in H3O+ or H7O3+ cations with asymmetric O–H⋯O groups.5,42 It follows from these data that the disappearance of the δH2O bands of all six H2O molecules in the H13O6+ cation is indicative of high symmetry of the central OHO group with a flat-bottomed H+ potential function and distribution of its dynamic properties over the 4 neighboring H-bonds. Breaking the symmetry should result in the reappearance of these bands.
Consistent with this, the δH2O band reappears for peripheral water molecules of the H13O6+ cation when they are shared with the solvation shell of the anion. As discussed above with respect to ion pairing, shared water molecules have decreased proton mobility and the local symmetry of the cation is lowered.
Conclusions
The ionized proton of a strong acid in liquid water, H+(aq), is an entirely unique species. On the IR timescale, a single species exists having constant composition, H+(H2O)6, and a well-defined boundary of positive charge delocalization. Suggestions of an equilibrium between two types of cations such as Eigen-type H3O·3H2O+ or Zundel-type H5O2+ ions10–15,75,76 are not supported by experiment.The distinctive IR spectrum of H+(aq), which is a combination of a continuous broad absorption (cba) and a relatively simple line spectrum, is not found in any gas phase H+(H2O)n cluster, even up to n > 100.28 This may be because in gas phase H+(H2O)n clusters, the 3D network structure promoted by a single H+ extends over the whole cluster network up to n = 100 (a “nucleation” effect)28 whereas in bulk water the excess proton changes the H-bonding network only up to n = 6. In addition, the excess proton locates itself near the surface of the gas-phase cluster23,25,26,77–81 where the environment has lower symmetry and where proton mobility is lower than in bulk water. For want of a better term, these gas phase clusters experience a microscopic “surface tension” that makes their environment different from bulk water. For the same reasons, i.e. lower symmetry and lower proton mobility, the structures of H+(H2O)n clusters in organic solvents and in crystals are distinctly different from those in liquid water. The only exception is found in crystals of H(CHB11I11)·8H2O48 where columns of centrosymmetrically located H+(H2O)6 cations find themselves in a nanotubular environment akin to liquid water.
The uniqueness of H13O6+(aq) lies in the unusual degree of delocalization of positive charge away from the central O–H–O group. Unlike typical Zundel-type H5O2+ cations, there is an unusually effective redistribution of charge onto peripheral parts of the H13O6+ cation and even out onto the O-atoms of eight outer-sphere water molecules not included in the n = 6 formulation. This results in a marked elongation of the central OHO group from ∼2.40 Å in Zundel-type ions to ∼2.57 Å in the H13O6+(aq) ion. There is concomitant lengthening in the four contiguous OHO groups from ∼2.52 Å in the Zundel-type H5O2·4H2O structure19 to ∼2.7 Å in H13O6+(aq). The H13O6+(aq) cation is not representative of an aquated Zundel-like H5O2+ cation. Rather, it is a unique H13O6+(aq) entity existing only in liquid water or in an environment similar to liquid water.
The unprecedented long O⋯O distance of ∼2.57 Å for the central O–H–O group in H13O6+(aq) cation raises a number of interesting questions. Where is the central H-atom located? On the IR timescale, the data indicate a symmetric distribution but cannot distinguish between a predominant mid-point location and a time-averaged distribution of asymmetric H-bonds with a nearly flat-bottomed, double-well H+ potential. Calculations at shorter timescales favor the predominance of an asymmetric structure29,30 but these calculations are done with short O⋯O distances typical of less delocalized cations, quite unlike the ∼2.57 Å indicated by the present experiments.
Another issue is the origin of the cba. Is there a connection between the unusual elongation of the central O–H–O group and the appearance of the cba, a feature found only in IR spectrum of H+(aq) clusters? This phenomenon is clearly associated with the fast dynamics of protons in these clusters and a challenge for theory to reproduce. The finding that at room temperature ∼30% of the IR spectrum of H+(aq) appears as a line spectrum whereas the remaining ∼70% appears as cba provides a critical test of the veracity of, or calibration for, such calculations.
A related problem is to understand the disappearance of the δH2O band in the line spectrum of H+(aq) clusters, as well as the νCO and δCOH bands of H+(CH3OH)n clusters64 and related bands in proton disolvates H+L2.58,61,65,66 This phenomenon is clearly related to the proximity of atoms to low-barrier H-bonds whose rapid fluctuations apparently lead to dramatically diminished intensities of associated vibrations.
Finally, we comment on the prevalence of ion pairing, even in the strongest acids. This may come as a surprise to many readers since strong acids are typically viewed in terms complete ionization, without ion pairing, and are treated as ideal electrolytes. Ionized strong acids can be considered as contact ion pairs. The H13O6+ cation has contact with the first hydration shell of the anion and they are not separated by bulk water molecules. Carborane acids show a slight dependence of their IR spectra on the nature of the carborane anion47 which is assigned to ion pairing effects (Scheme 3). With weaker acids such as HCl, or increasingly concentrated solutions of strong acids such as HNO3 and HClO4, one or two peripheral water molecules of the n = 6 cation can be shared with the first solvation of the anion in contact ion pairs with reduced overall water content (Scheme 2). The influence on the IR spectrum is mainly in changes in band intensities. Our results obtained for dilute HCl solutions do not contradict those of Agmon82 in highly concentrated HCl solutions (≥8 M) where a Zundel-type H5O2+·4H2O ion surrounded by Cl-anions and a short central 2.40 Å O⋯O distance was established. This proposed structure is very similar to that found by Marsh et al. in the crystal state19 and can be viewed as a H5O2+ cation in contact with the first hydration shells of neighboring Claq− anions. This removes the unique properties of the aqueous H13O6+ cation and the core reverts to the common Zundel-type H5O2+ cation.
Ion pairing is the underlying reason why the structure of the H+(H2O)6 cation in aqueous solution changes to H7O3+·nH2O in weakly basic organic solvents such as benzene and chlorinated hydrocarbons.42 In organic solvents of low permittivity, diminished hydration of the anion leads to the formation of more intimate ion pairs with the hydrated proton. The stronger polarizing influence of the anion on the hydrated proton distorts its core to a H7O3+ cation. Tighter ion pairing suppresses positive charge delocalization so unlike the H13O6+ cation in aqueous solution, the O⋯O distances are typical of a static hydrated H7O3+ cation.
This phenomenon plays out in a parallel manner in the nanotubular structure of H(CHB11I11)·8H2O, but with an important difference. H(H2O)6+ clusters located in the center of the tube adopt the long O⋯·O bonded, centrosymmetric H13O6+ structure found in aqueous solution whereas those near the anionic walls of the tube have a distorted core that approaches the H7O3+ motif. Notably, the core of this ion has O⋯O distances that are unusually long (∼2.62 Å)48 compared to those of typical H7O3+·nH2O cations in crystals or organic solutions (∼2.50 Å).42 Just as the longer O⋯O bonded central H5O2 group of the centrosymmetric H13O6+ ion retains topographical similarity to H5O2+, so this H7O3 group with longer bond lengths retains a topographical similarity to the H7O3+ ion. The difference lies in the surrounding environment. H(H2O)6+ clusters in the nanotubular structure are more hydrated and less tightly ion paired than the shorter O⋯·O bonded H7O3+·nH2O clusters in crystals or organic solvents because the positive charge is more delocalized. The tube environment is more akin to liquid water.
This is the sine qua non of our findings. Liquid water provides a special environment for H+ that leads to greater delocalization of positive charge than previously suspected.
Acknowledgements
This work was supported by NIH Grant GM023851.Notes and references
- S. Cukierman, Biochim. Biophys. Acta, Bioenerg., 2006, 1756, 876 CrossRef.
- C. J. T. Grotthuss, Ann. Chim., 1806, LVIII, 54 Search PubMed.
- L. J-Gay-Lussac and A. Humboldt, Ann. Chim., 1805, LIII, 239 Search PubMed.
- M. Eigen, Angew. Chem., Int. Ed. Engl., 1964, 3, 1 CrossRef.
- E. S. Stoyanov, C. K-Kim and C. A. Reed, J. Am. Chem. Soc., 2006, 128, 1948 CrossRef CAS.
- O. F. Mohammed, D. Pines, J. Dreyer, E. Pines and E. T. J. Nibbering, Science, 2005, 310, 83 CrossRef CAS.
- O. F. Mohammed, D. Pines, E. Pines and E. T. J. Nibbering, Chem. Phys., 2007, 341, 240 CrossRef CAS.
- G. Zundel in The Hydrogen Bond: Recent Developments in Theory and Experiments, Schuster, P.; Zundel, G.; Sandorfy, C.; ed.; North-Holland: Amsterdam, 1976; Vol. 2 Search PubMed.
- Hydrogen Bond (Vodorodnaya Svias'); Ed.: N. D. Sokolov, Moscow, Nauka, 1981 Search PubMed.
- J. Kim, U. W. Schmitt, J. A. Gruetzmacher, G. A. Voth and N. E. Scherer, J. Chem. Phys., 2002, 116, 737 CrossRef CAS.
- R. Vuilleumier and D. Borgis, J. Chem. Phys., 1999, 8, 3116.
- J. M. Heuft and E. J. Meijer, Phys. Chem. Chem. Phys., 2006, 35, 3523 Search PubMed.
- S. Woutersen and H. J. Bakker, Phys. Rev. Lett., 2006, 96, 138305 CrossRef.
- A. Botti, F. Bruni and M. A. Ricci, J. Chem. Phys., 2006, 125, 014508 CrossRef CAS.
- G. A. Voth, Acc. Chem. Res., 2006, 39, 143 CrossRef CAS.
- A. Likholyot, K. H. Lemke, J. K. Hovey and T. M. Seward, Geochim. Cosmochim. Acta, 2007, 71, 2436 CrossRef CAS.
- B. Krebs, S. Bonmann and K. Erpenstein, Z. Naturforsch., 1991, 468, 919 Search PubMed.
- Z. Xie, R. Bau and C. A. Reed, Inorg. Chem., 1995, 34, 5403 CrossRef CAS.
- R. Bell, G. Christopher, F. R. Fronczek and R. E. Marsh, Science, 1975, 190, 151 CAS.
- M. Okumura, L. I. Yeh, J. D. Myers and Y. T. Lee, J. Phys. Chem., 1990, 94, 3416 CrossRef CAS.
- J.-C. Jiang, Y.-S. Wang, H.-C. Chang, S. H. Lin, Y. T. Lee and G. Niedner-Schatteburg, J. Am. Chem. Soc., 2000, 122, 1398 CrossRef CAS.
- M. Miyazaki, A. Fujii, T. Ebata and N. Mikami, Science, 2004, 304, 1134 CrossRef CAS.
- J.-W. Shin, N. I. Hammer, E. G. Diken, M. A. Johnson, R. S. Walters, T. D. Jaeger, M. A. Duncan, R. A. Christie and K. D Jordan, Science, 2004, 304, 1134 CrossRef CAS.
- C.-K. Lin, C.-C. Wu, Y.-S. Wang, Y. T. Lee, H.-C. Chang, J.-L. Kuo and M. L. Klein, Phys. Chem. Chem. Phys., 2005, 7, 938 RSC.
- H.-C. Chang, C.-C. Wu and J.-L. Kuo, Int. Rev. Phys. Chem., 2005, 24, 553 CrossRef CAS.
- C.-C. Wu, C.-K. Lin, H.-C. Chang, J.-C. Jiang, J.-L. Kuo and M. L. Klein, J. Chem. Phys., 2005, 122, 074315 CrossRef.
- J. M. Headrick, E. G. Diken, R. S. Walters, N. I. Hammer, R. A. Christie, J. Cui, E. M. Myshakin, M. A. Duncan, M. A. Johnson and K. D. Jordan, Science, 2005, 308, 1765 CrossRef CAS.
- K. Mizuse, A. Fujii and N. Mikami, J. Chem. Phys., 2007, 126, 231101 CrossRef.
- M. Tuckerman, K. Laasonen, M. Sprik and M. Parrinello, J. Chem. Phys., 1995, 103, 150 CrossRef CAS.
- O. Markovitch, H. Chen, S. Izvekov, F. Paesani, G. Voth and N. Agmon, J. Phys. Chem. B, 2008, 112, 9456 CrossRef CAS.
- G. Zundel and M. Eckert, J. Mol. Struct., 1989, 200, 73 CrossRef.
- S. Bratos and H. F. Ratajczak, Chem. Phys., 1982, 73, 77.
- H. Romanovsky and L. Sobezek, Chem. Phys., 1977, 19, 361 CrossRef.
- N. B. Librovich, V. P. Sakun and N. D. Sokolov, Chem. Phys., 1979, 39, 351 CrossRef CAS.
- N. B. Librovich, V. P. Sakun and N. D. Sokolov, In: Vodorodnaya Svias' (Hydrogen Bond), Moscow, Nauka, 1981, p.174 Search PubMed.
- G. L. Hofacker, Y. Marechal and M. A. Ratner, In: The Hydrogen Bond. Recent Developments in Theory and Experiments. ed.: P. Schuster, G. Zundel and C. Sandorfy. V.1, North-Holland, Amsterdam, 1976 Search PubMed.
- G. V. Yukhnevich and E. G. Tarakanova, J. Mol. Struct., 1988, 177, 495 CrossRef.
- G. V. Yukhnevich, E. G. Tarakanova, V. D. Mayorov and N. B. Librovich, J. Mol. Struct., 1992, 265, 234 CrossRef.
- R. Vuilleumier and D. Borgis, J. Chem. Phys., 1999, 111, 4251 CrossRef CAS.
- K. E. Laasonen and M. L. Klein, J. Phys. Chem. A, 1997, 101, 98 CrossRef CAS.
- A. J. Sillanpaa and K. E. Laasonen, Phys. Chem. Chem. Phys., 2004, 6, 555 RSC.
- E. S. Stoyanov, I. V. Stoyanova, F. S. Tham and C. A. Reed, J. Am. Chem. Soc., 2008, 130, 12128 CrossRef CAS.
- K. Rahmelow and W. Hübner, Appl. Spectrosc., 1997, 51, 160 CrossRef CAS.
- J.-J. Max and C. Chapados, Appl. Spectrosc., 1998, 52, 963 CrossRef CAS.
- E. S. Stoyanov, J. Chem. Soc., Faraday Trans., 1998, 94, 2803 RSC.
- E. S. Stoyanov, Phys. Chem. Chem. Phys., 1999, 1, 2961 RSC.
- E. S. Stoyanov, I. V. Stoyanova and C. A. Reed, J. Am. Chem. Soc., 2010, 132, 1484 CrossRef CAS.
- E. S. Stoyanov, I. V. Stoyanova, F. S. Tham and C. A. Reed, J. Am. Chem. Soc., 2009, 131, 17540 CrossRef CAS.
- Y. Maréchal, J. Chem. Phys., 1991, 95, 5565 CrossRef CAS.
- J. E. Bertie and Z. Lan, Appl. Spectrosc., 1996, 50, 1047 CAS.
- S. Nunn and K. Nishikida, Application Note: 50581: Advanced ATR Correction Algorithm, Thermo Fisher Scientific Inc. Madison, WI, USA, 2008 (http://www.thermo.com/eThermo/CMA/PDFs/Articles/articlesFile_7217.pdf) Search PubMed.
- J. Stangret and J. Gampe, J. Phys. Chem. A, 2002, 106, 5393 CrossRef CAS.
- A. W. Omta, M. F. Kropman, S. Woutersen and H. J. Bakker, Science, 2003, 301, 347 CrossRef CAS.
- J. D. Smith, R. J. Saykally and P. L. Geissler, J. Am. Chem. Soc., 2007, 129, 13847 CrossRef CAS.
- E. S. Stoyanov, C. K-Kim and C. A. Reed, J. Am. Chem. Soc., 2006, 128, 8500 CrossRef CAS.
- For English version, see Handbook of Chemist (Spravochnik Himika), Ed.: B. P. Nikol'skii, Moscow-Leningrad, 1964, Vol. 3, 2nd edition (Russian). http://molbiol.ru/eng/protocol/01_23.html Search PubMed.
- E. S. Stoyanov, J. Chem. Soc., Faraday Trans., 1997, 93, 4165 RSC.
- E. S. Stoyanov and C. A. Reed, J. Phys. Chem. A, 2006, 110, 12992 CrossRef CAS . Note: While band frequencies often decrease with increasing bond length, especially those arising from symmetric vibrations, the Zundel ion marker band is an asymmetric stretch and empirically follows an inverse relationship to bond length.
- N. I. Hammer, E. G. Diken, J. R. Roscioli, M. A. Johnson, E. M. Myshakin, K. D. Jordan, A. B. McCoy, X. Huang, J. M. Bowman and S. Carter, J. Chem. Phys., 2005, 122, 244301 CrossRef.
- J. R. Roscioli, L. R. McCunn and M. A. Johnson, Science, 2007, 316, 249 CrossRef CAS.
- E. S. Stoyanov and I. V. Smirnov, J. Mol. Struct., 2005, 740, 9 CrossRef CAS.
- M. F. Kropman and H. J. Bakker, Science, 2001, 291, 2118 CrossRef CAS.
- M. Juhasz, S. Hoffmann, E. Stoyanov, K.-C. Kim and C. A. Reed, Angew. Chem., Int. Ed., 2004, 43, 5352 CrossRef CAS.
- E. S. Stoyanov, I. V. Stoyanova and C. A. Reed, Chem.–Eur. J., 2008, 14, 3596 CrossRef CAS.
- E. S. Stoyanov, Mendeleev Commun., 1999, 190 RSC.
- E. S. Stoyanov, Phys. Chem. Chem. Phys., 2000, 2, 1137 RSC.
- M. Smiechowsky and J. Stangret, J. Mol. Struct., 2008, 878, 104 CrossRef.
- D. Hadzi and N. Kobilarov, J. Chem. Soc. A, 1966, 439 RSC.
- H. Kanno and J. Hiraishi, Chem. Phys. Lett., 1984, 107, 438 CrossRef CAS.
- D. Schiöberg and G. Zundel, Chem. Phys. Lett., 1976, 38, 334 CrossRef.
- L. I. Katzin, G. W. Mason and D. F. Peppard, Spectrochim. Acta, 1978, 34A, 51 CrossRef CAS.
- E. Gebert, A. H. Reis, S. W. Peterson, L. I. Katzin, G. W. Mason and D. F. Peppard, J. Inorg. Nucl. Chem., 1981, 43, 1451 CAS.
- D. Hadzi and A. Novak, Proc. Chem. Soc., 1960, 241 Search PubMed.
- M. Callery and J. C. Speakman, Acta Crystallogr., 1964, 17, 1097 CrossRef.
- D. Marx, M. E. Tuckerman, J. Hutter and M. Parrinello, Nature, 1999, 397, 601 CrossRef CAS.
- M. Cavalleri, L.-Å. Näslund, D. C. Edwards, P. Wernet, H. Ogasawara, S. Myneni, L. Ojamäe, M. Odelius, A. Nilsson and L. G. M. Pettersson, J. Chem. Phys., 2006, 124, 194508 CrossRef.
- J.-L. Kuo and M. L. Klein, J. Chem. Phys., 2005, 122, 024516 CrossRef.
- T. James and D. J. Wales, J. Chem. Phys., 2005, 122, 134306 CrossRef.
- S. S. Iyengar, M. K. Petersen, T. J. F. Day, C. J. Burnham, V. E. Teige and G. A. Voth, J. Chem. Phys., 2005, 123, 084309 CrossRef.
- C. J. Burnham, M. K. Petersen, T. J. F. Day, S. S. Iyengar and G. A. Voth, J. Chem. Phys., 2006, 124, 024327 CrossRef.
- N. J. Singh, M. Park, S. K. Min, S. B. Suh and K. S. Kim, Angew. Chem., Int. Ed., 2006, 45, 3795 CrossRef CAS.
- N. Agmon, J. Phys. Chem. A, 1998, 102, 192 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: experimental details, IR spectra. See DOI: 10.1039/c0sc00415d |
| This journal is © The Royal Society of Chemistry 2011 |