Convergent 18F radiosynthesis: A new dimension for radiolabelling†‡
Lei
Li
a,
Matthew N.
Hopkinson
a,
Rodrigue Leuma
Yona
a,
Romain
Bejot§
a,
Antony D.
Gee
b and
Véronique
Gouverneur
*a
aUniversity of Oxford, Chemistry Research Laboratory, 12 Mansfield Road, OX1 3TA, Oxford, UK. E-mail: veronique.gouverneur@chem.ox.ac.uk
bADD: Division of Imaging Sciences and Bioengineering, King’s College London, St Thomas’ Hospital, London, SE1 7EH, UK
First published on 1st October 2010
Abstract
The availability of radiolabelled probes is important for in vivo studies by positron emission tomography (PET). Among the frontier challenges in 18F-radiochemistry are the interconnected goals of increasing synthetic efficiency and diversity in the construction of 18F-labelled radiotracers. 18F-Radioretrosynthetic strategies implemented to date are typically linear sequences of transformations designed with the aim of introducing the 18F-label ideally in the last step or at least as late as possible. Here we report that convergent 18F-radiochemistry allows for the rapid assembly of functionalised 18F-radiotracers from readily accessible 18F-labelled prosthetic groups. Using multicomponent reactions for proof of concept, Ugi-4CR, Passerini-3CR, Biginelli-3CR and Groebke-3CR were performed successfully using 18F-benzaldehydes and these highly convergent reactions delivered, in high radiochemical yield (RCY), structurally complex 18F-radiotracers with the label positioned on an aryl motif not responsive to direct nucleophilic fluorination. These data establish an unprecedented connection between radiochemistry for PET and the field of multicomponent chemistry and demonstrate that convergent retroradiosynthesis is a powerful strategy expanding dramatically the scope of 18F-prosthetic group radiochemistry. The preparation of 18F-labelled prosthetic groups from [18F]fluoride ion is commonly performed in many labelling laboratories, so the concept of convergent 18F-radiosynthesis can easily be applied immediately.
Introduction
Positron emission tomography (PET) is a non-invasive molecular imaging technique, which relies on the availability of radiolabelled molecular probes for molecular-level diagnostics, biological research and drug discovery.1 Worldwide, cyclotron sites are in place for the production of radioisotopes and selected labelled biomarkers, but to date, rapid progress in PET imaging is limited by the cost, speed, and efficiency of synthetic methods to access structurally diverse radiolabelled probes. Ideally, the radiosynthetic protocol should be as efficient as possible to minimise functional manipulation after introduction of radioisotopes and to avoid the formation of side-products that cause complications during purification.218F is commonly identified as the radionuclide of choice due, in part, to its relatively long half-life of 110 min which allows for multistep radiosynthetic protocols. In addition, the low emitted positron energy of fluorine-18 can allow for higher resolution PET images.3 Direct 18F-fluorination is dominated by [18F]fluoride ion chemistry for two main reasons; this [18F]fluorine source is much easier to access and handle than [18F]F2, and the resulting 18F-labelled probes are produced in higher specific radioactivity, a key feature allowing for true tracer methods in PET.4 This strategy however suffers from limitations. [18F]fluoride ion based radiochemistry is relatively limited in scope. The one element of structure that invariably increases the difficulty of a radiosynthesis is the presence in a complex 18F-probe of a fluoroaromatic motif not accessible upon direct nucleophilic aromatic substitution. This is one of the frontier challenges in 18F-radiochemistry. Moreover, the harsh conditions required for successful nucleophilic 18F-fluorination are not compatible for the labelling of biological molecules. Indirect 18F-labelling is a method based on the biomolecular coupling of a small 18F-labelled reactive precursor with the probe of interest. This strategy is very often used for the labelling of peptides or to access structural motifs not accessible by direct nucleophilic 18F-fluorination. Independent of the chosen strategy (direct or indirect 18F-labelling), the challenge is for the total radiosynthesis time (synthesis, purification, analysis and formulation) not to exceed roughly three isotope's half-lives (∼ 6 h).5An analysis of the state of play of 18F-radiochemistry for PET reveals two intriguing characteristics. Radiochemists often select the site of 18F-radiolabelling to fit existing radiosynthetic methods, an approach that could be detrimental for rapid progress in PET probe discovery. In addition, we noted that the 18F-radioretrosynthetic strategies applied to date are typically based on linear sequences of transformations designed with the aim of introducing the 18F-label ideally in the last step or at least as late as possible. This criteria becomes increasingly difficult to meet when the probe is structurally complex or highly functionalised. The central proposition advanced herein is that 18F-radiochemistry may benefit from convergent synthetic tactics that should allow for the rapid assembly of 18F-radiotracers from a readily accessible 18F-labelled prosthetic group. Accordingly, radiochemists will have the possibility to introduce the 18F-label on different positions of the target probe without the need to redesign the overall radiosynthetic route.
To validate this new conceptual framework, we selected multicomponent reactions (MCR) because these highly convergent atom- and step-economic procedures can deliver structurally diverse molecules in a single step by reacting three or more substrates simultaneously.6,7 This class of reactions is attractive because the 18F-labelled precursor becomes integrated in the intrinsic structure of the probe. Despite the extensive use of MCRs in organic synthesis and medicinal chemistry, radiochemists have not explored the potential of the MCRs in the context of 18F-indirect labelling. Herein, we report the first so-called radio-MCRs with 18F-labelled components and illustrate the value of this approach with the preparation of complex 18F-labelled compounds that are not accessible by direct nucleophilic fluorination. This study establishes that convergent radiosynthesis is a powerful strategy expanding dramatically the scope of 18F-prosthetic group radiochemistry. The method is simple, which is a clear advantage for radiochemistry, yet its ingenuity allows for the synthesis of structurally diverse 18F-labelled molecular scaffolds.
The 18F-labelling of aromatics not activated with electron-withdrawing groups remains notoriously difficult to perform with cyclotron-produced [18F]fluoride ion, the preferred source of fluorine since it is available in high specific activity.4 Diaryliodonium salts are suitable precursors to access 18F-labelled electron-rich aromatics with [18F]fluoride but the structural complexity of the [18F]products is relatively limited.8 As proof of concept, we therefore chose to validate this novel MCR-based 18F-radiochemistry with the view to synthesise 18F-labelled compounds that are not obvious candidates for a retro-radiosynthetic route based on nucleophilic 18F-fluorination. 4-[18F]fluorobenzaldehyde and substituted derivatives were selected as radioactive components because aldehydic components are commonly used in many high profile “cold” MCRs.9 The preparation of 4-[18F]fluorobenzaldehyde from [18F]fluoride ion is well documented in the literature.10Fig. 1 illustrates the potential of this approach with the preparation of 3,4-dihydropyrimidin-2-(1H)-one with the 18F-label at the aryl group. Upon radio-MCR, the readily accessible 18F-benzaldehyde will deliver an 18F-labelled 3,4-dihydropyrimidin-2-(1H)-one, a compound difficult to access using [18F]fluoride ion as the necessary precursor would feature an aryl group not sufficiently activated for classical SNAr (Fig. 1).
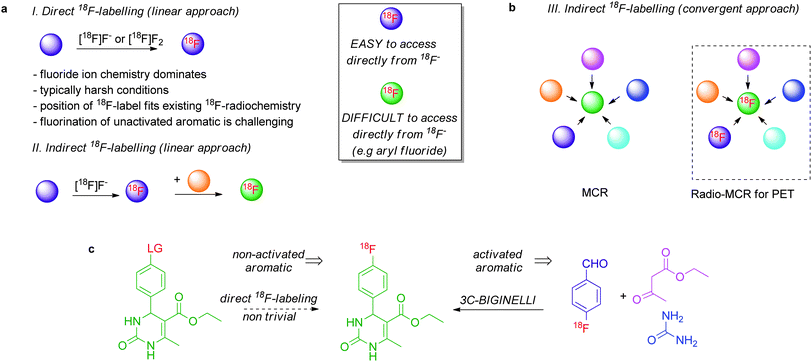 | ||
| Fig. 1 Convergent Radiosynthesis: radio-MCR with 18F-Labelled Building Blocks. a. linear radiosynthesis to 18F-labelling; b. a convergent retro-radiosynthetic approach to access 18F-radiotracers not accessible upon direct 18F-fluorination; c. MCR Biginelli reaction with 18F-labelled benzaldehyde; LG = leaving group. | ||
Results and discussion
The use of fluorine-containing building blocks in MCRs is relatively limited despite the popularity of fluorine in medicinal chemistry.7,11 In preliminary work, we validated the feasibility of four representative MCRs using the fluorinated aldehydes 1a or 1b. Upon cyclocondensation of 1a or 1b with ethyl acetoacetate and urea in acetonitrile, the Biginelli reaction, one of the oldest and most studied MCRs,12 gave, after 5 h at 100 °C, the 3,4-dihydropyrimidin-2-(1H)-ones132a and 2b in 80% and 90% yield, respectively. Three isocyanide-based MCRs (IMCRs) were examined next. IMCRs have recently attracted a lot of interest since they are compatible with a range of functional groups not involved in the initial MCR. This characteristic allows for multiple post-condensation transformations leading to products of extraordinary structural complexity. The four-component Ugi reaction was performed with 4-fluorobenzaldehyde 1a to give the peptidic-like products 3a and 3b in good yields. At room temperature, these transformations were completed within 48 h in methanol. The fluorinated imidazo[1,2-a]pyridines 4a and 4b were made available using the three-component Groebke-Bienaymé-Blackburn reaction.14 This MCR is important in the context of drug discovery as related products have found applications across a wide range of therapeutic areas and for PET imaging.15 The α-acyloxyamides 5a–d were obtained in up to 98% yield, condensing 1a with cyclohexylisocyanide and various carboxylic acids. These Passerini reactions16 were performed at room temperature and completed within 8 h (Fig. 2).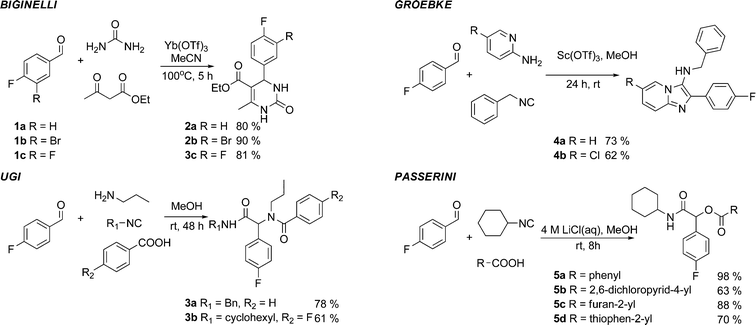 | ||
| Fig. 2 MCR with Fluorinated Benzaldehydes 1a-b. | ||
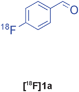
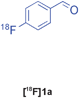
The radiolabelled components selected for this study are 4-[18F]fluorobenzaldehyde [18F]1a, its halide substituted derivatives [18F]1b-c and [18F]4-fluorobenzoic acid [18F]6 (Table 1). The 18F-components [18F]1a-b were prepared in good RCY by fluorination (SNAr) of the corresponding 4-trimethylammonium aryl triflates using [18F]KF/Kryptofix® 222 (Table 1, entries 1–2). The synthesis of [18F]1c applying this conventional fluorination protocol was not successful, possibly because of the thermal instability of the precursor 7c upon extended reaction times (condition B). Pleasingly, the radiofluorination of 7c took place when performed on a microfluidic reaction chip17 at 200 °C for 4 min. Under these conditions, [18F]1c was formed in 73% RCY (Table 1, entry 3), a result highlighting the advantage of micro-reactor technology over traditional systems in the context of radiolabelling. The synthesis of [18F]6 was performed within 20 min upon nucleophilic radiofluorination followed by hydrolysis (Table 1, entry 4). The specific activities18 of 4-[18F]fluorobenzaldehyde and 3,4-[4-18F]difluorobenzaldehyde were determined to be 35–90 GBq μmol−1 (n = 3) and 30–50 GBq μmol−1 (n = 3) decay-corrected to the end of synthesis respectively, starting from 200–500 MBq of [18F]fluoride ion. The 18F-components (5–15 MBq) were purified by solid-phase extraction (SPE) and immediately subjected to multicomponent condensation.

| Entry | 18F-component | Conditionsa | RCY [%]b | n | S. A.c(GBq μmol−1) |
|---|---|---|---|---|---|
| a Conditions A. 7a (10 mg), DMSO (0.5 mL), microwave 50 W, 100 s; Conditions B. 7b (10 mg), MeCN (0.3 mL), 120 °C, 10 min; Conditions C. 7c (2 mg), MeCN (40 μL), 200 °C, 4 min; Conditions D. 8 (10 mg), MeCN (0.3 mL), 120 °C, 15 min followed by nPr4NOH (aq. 1 M, 200 μL), 120 °C, 5 min then CF3COOH (aq. 1 M, 200 μL), rt, 5 min. b Crude RCY based on n experiments (decay-corrected). c S. A.: Specific Activity to the end of synthesis. | |||||
| 1 | [18F]1a | A | 83 | 3 | 35–90 (n = 3) |
| 2 | [18F]1b | B | 90 | 1 | — |
| 3 | [18F]1c | C | 73 | 3 | 30–59 (n = 3) |
| 4 | [18F]6 | D | 56 | 2 | — |
With the 18F-labelled aldehydes in hand, we were poised to investigate whether the convergent radio-MCRs would proceed as planned. The use of 18F-components in radio-MCRs raises fundamental questions. It is well documented that the efficiency of MCRs is dependent on various parameters such as temperature, concentration, pressure, reaction stoichiometry and the nature and the order of addition of the components.19 Our preliminary work indicates that the conditions typically used for MCRs (reaction time from 5 h to 48 h) are not compatible with the short half-life of 18F. Moreover, the 18F-labelled component will be present in severe sub-stoichiometric amount (pmol-nmol) in comparison with the non-labelled components (μmol-mmol).
These considerations led to extensive optimisation studies for each MCR as detailed for the representative Groebke-Bienaymé-Blackburn reaction in Table 2. In order to reduce the reaction time, it was necessary to change the solvent from the low-boiling methanol used in the ‘cold’ process. With acetonitrile as solvent, the desired 18F-labelled imidazo[1,2-a]pyridine product was formed but in poor RCY (< 10%, Table 2, entries 1–2). Switching to the polar protic 3-methyl-1-butanol afforded [18F]4a in 31% RCY when heated to 110 °C for 15 min (Table 2, entry 3). Increasing the reaction temperature to 150 °C led to a radiochemical yield of 73% which was improved to 81% when microwave irradiation (50W) was used as the heating method (Table 2, entries 4–5). The order of addition of the reagents was found to affect the reaction efficiency. When the isocyanide component was added prior to the radioactive benzaldehyde, a decrease in the radiochemical yield to 64% was observed (Table 2, entry 7). The RCY of [18F]4a could be increased to 85% (n = 4) when the reaction was heated to 170 °C under conventional heating for 15 min (Table 2, entry 8).
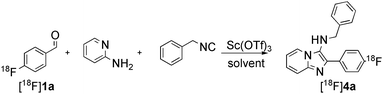
| Entrya | Temp./°Cb | Time/min | Solvent | RCY (%)c |
|---|---|---|---|---|
| a A solution of [18F]1a in the solvent (200 μL) was dispensed into a vial containing 2-aminopyridine (3 mg) and Sc(OTf)3 (5 mg). Benzyl isocyanide (3 μL) in the solvent (300 μL) was then added via syringe. b Conventional heating. c Crude RCY determined by radio-TLC (decay-corrected). d 100 °C for 15 min followed by 5 min at 120 °C. e Performed under microwave irradiation (50 W). f Benzyl isocyanide added prior to [18F]4-fluorobenzaldehyde. | ||||
| 1 | 100 | 15 | MeCN | 3 |
| 2 | 100–120d | 20 | MeCN | 8 |
| 3 | 110 | 15 | 3-Methyl-1-butanol | 31 |
| 4 | 150 | 15 | 3-Methyl-1-butanol | 73 |
| 5 | 150e | 15 | 3-Methyl-1-butanol | 81 |
| 6 | 150 | 30 | 3-Methyl-1-butanol | 80 |
| 7 | 150f | 15 | 3-Methyl-1-butanol | 64 |
| 8 | 170 | 15 | 3-Methyl-1-butanol | 85 (n = 4) |
Our results for the radio-Biginelli, -Ugi, -Groebke and -Passerini reactions are summarised in Table 3. Details of the optimisation studies for all MCRs are included in the ESI.† The Yb(OTf)3-mediated condensation of [18F]benzaldehyde 1a with ethyl acetoacetate and urea in 3-methyl-1-butanol gave the dihydropyrimidinone [18F]2a in 66% RCY (n = 3) after 30 min at 130 °C (Table 3, entry 1). The brominated [18F]benzaldehyde 1b also successfully reacted under the same conditions, affording the corresponding radiolabelled Biginelli product [18F]2b in 38% RCY (Table 3, entry 2). [18F]2c, bearing two fluorine substituents, was delivered in 25% RCY (n = 3) upon reacting the 18F-labelled aldehyde 1c with urea and 4-methoxy-3-oxobutanoate (Table 3, entry 3). The four-component Ugi reactions were successfully performed using ethanol as solvent. [18F]1a was incorporated into the peptide-like compound [18F]3a in 62% RCY (n = 3) upon treatment with benzoic acid (6 mg), 1-propylamine (4 μL) and benzyl isocyanide (6 μL) in ethanol (200 μL) at 100 °C for 30 min (Table 3, entry 4). The efficiency of this transformation was found to be highly dependent on the reagent concentrations. Thus, performing the reaction under the same conditions but with half the reagent amounts in 500 μL of ethanol led to only a 7% conversion to the product. [18Fal]3b was successfully synthesised in 56% RCY (n = 2) upon treating [18F]1a with 4-fluorobenzoic acid, 1-propylamine and cyclohexyl isocyanide under optimised radio-Ugi conditions (Table 3, entry 5). This substrate, bearing two remote fluorine substituents, was selected to assess the possibility of labelling these Ugi products at an alternative position by using a different 18F-component. When the carboxylic acid [18F]6 was condensed under similar conditions with benzaldehyde, 1-propylamine and cyclohexyl isocyanide, no desired product was formed. However, the reaction took place affording [18Fac]3b in 10% RCY (n = 3) when the reaction mixture was spiked with non-labelled fluorobenzoic acid (Table 3, entry 6). This result indicates that, in accordance with the optimization studies, this carboxylic acid component must be present at a higher concentration for the MCR to proceed. The radio-Groebke-Bienaymé-Blackburn reaction was a remarkably clean process. Under the optimised conditions described in Table 2, [18F]4a was delivered as the only radioactive product in 85% RCY (n = 4, Table 3, entry 7). The HPLC chromatogram for this reaction displaying the radioactivity and reference UV traces is shown in Fig. 3. The chlorine-substituted imidazo[1,2-a]pyridine [18F]4b, whose core bears similarities with that of the anxiolytic drug alpidem, was formed in 76% RCY (n = 3, Table 3, entry 8). The 4 M aqueous lithium chloride solution used as solvent for the ‘cold’ Passerini reactions was found to be incompatible with the 18F-labelled aldehydic component, which must be dissolved in an organic solvent during purification. Extensive optimisation studies identified a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 mixture of MeOH:LiCl(aq) as a suitable solvent system, delivering the Passerini product [18F]5a in 65% RCY (n = 3) upon heating [18F]1a, benzoic acid and cyclohexyl isocyanide at 100 °C for 30 min (Table 3, entry 9). The process was successfully applied to a variety of heteroaryl carboxylic acids affording the 2,5-dichloropyridine, furan and thiophene substituted products [18F]5b, [18F]5c and [18F]5d in 33% (n = 2), 28% (n = 2) and 32% (n = 2) RCY respectively (Table 3, entries 10–12).
:6 mixture of MeOH:LiCl(aq) as a suitable solvent system, delivering the Passerini product [18F]5a in 65% RCY (n = 3) upon heating [18F]1a, benzoic acid and cyclohexyl isocyanide at 100 °C for 30 min (Table 3, entry 9). The process was successfully applied to a variety of heteroaryl carboxylic acids affording the 2,5-dichloropyridine, furan and thiophene substituted products [18F]5b, [18F]5c and [18F]5d in 33% (n = 2), 28% (n = 2) and 32% (n = 2) RCY respectively (Table 3, entries 10–12).
| Entry | 18F-building block | 18F-labelled product | MCR | Labelling procedurea | RCY(%)b |
|---|---|---|---|---|---|
| a Conditions: A. urea (3 mg), β-ketoester (6 μL), Yb(OTf)3 (3 mg), 3-methyl-1-butanol (0.2 mL), 130 °C, 30 min; B. 1-propylamine (4 μL), isocyanide (6-7 μL), benzoic acid (8 mg), ethanol (0.2 mL), 100 °C, 30 min; C. 2-aminopyridine (3 mg), Sc(OTf)3 (5 mg), benzyl isocyanide (3 μL) in 3-methyl-1-butanol (0.5 mL), 170 °C 15 min; D. carboxylic acid (7 mg), cyclohexyl isocyanide (4 μL) in 4M LiCl (aq., 600 μL), methanol (0.1 mL), 100 °C, 30 min. b Crude RCY based on n experiments (decay-corrected). c Reaction spiked with ‘cold’ 4-fluorobenzoic acid (8 mg). | |||||
| 1 |

|

|
Biginelli | A | 66(n = 3) |
| 2 |

|

|
Biginelli | A | 38(n = 1) |
| 3 |

|

|
Biginelli | A | 25(n = 3) |
| 4 |

|

|
Ugi | B | 62(n = 3) |
| 5 |

|
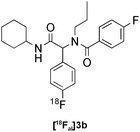
|
Ugi | B | 56(n = 2) |
| 6c |

|
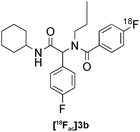
|
Ugi | B | 10(n = 2) |
| 7 |

|

|
Groebke | C | 85(n = 4) |
| 8 |

|

|
Groebke | C | 76(n = 3) |
| 9 |

|

|
Passerini | D | 65(n = 3) |
| 10 |
|

|
Passerini | D | 33(n = 2) |
| 11 |
|

|
Passerini | D | 28(n = 2) |
| 12 |

|

|
Passerini | D | 32(n = 2) |
![HPLC
Chromatogram for the Radiosynthesis of [18F]4a.](/image/article/2011/SC/c0sc00362j/c0sc00362j-f3.gif) | ||
| Fig. 3 HPLC Chromatogram for the Radiosynthesis of [18F]4a. | ||
Currently, numerous pharmaceutical companies are investing in preclinical and clinical imaging centres to allow for the inclusion of imaging endpoints throughout drug development. 18F-Labelled small molecules can provide early clinical distribution data to facilitate compound selection or can offer valuable information by probing drug-receptor/enzyme occupancies and disease mechanisms across multiple therapeutic areas (e.g. CNS, oncology, metabolic and immunological disorders).20 MCRs have benefited medicinal chemists for many years allowing for rapid access to diversity-oriented libraries and facilitating high-throughput screening (HTS) for hit identification and efficient “hit-lead-drug” translation studies. MCR-based radiosynthetic strategy may therefore streamline the synthesis of a wide range of 18F-radiotracers and allow for rapid imaging studies of molecules derived from HTS. This approach is certainly advantageous over current practice that typically requires the development of a suitable radiosynthetic route after a lead compound has been selected for labelling. Having demonstrated the feasibility of radio-MCRs,21 our next objective was therefore to label a more complex drug-type target. For proof of concept, we selected compound 10 (L-771,668), a potent antagonist of the α1A-selective adrenoceptor.22 This compound is a functionalised dihydropyrimidinone accessible upon Biginelli condensation. The planned retro-radiosynthetic route to [18F]10 involves three steps, the preparation of the 18F-component, the Biginelli-3CR and a post-condensation event consisting of the attachment of the side chain. The 18F-component [18F]1c was condensed with urea and methyl 4-methoxy-3-oxobutanoate to give the dihydropyrimidinone [18F]2c in 25% RCY (n = 3, Table 3, entry 3). The coupling of the pre-activated side chain was successfully carried out in acetonitrile at 130 °C for 30 min. The overall sequence of reactions delivered the desired complex drug labelled on the benzene ring in just over one hour. The convergent nature of this concise radiosynthesis could offer access to various structural analogues by using alternative components (ketoester or urea) or upon attachment of other side chains (Fig. 4).
![Convergent Radiosynthesis of [18F]10.](/image/article/2011/SC/c0sc00362j/c0sc00362j-f4.gif) | ||
| Fig. 4 Convergent Radiosynthesis of [18F]10. | ||
Conclusions
In conclusion, we have demonstrated that multicomponent reactions are suitable for the preparation of 18F-labelled material. Using a MCR as the key transformation, an extremely concise two-step synthetic sequence for the synthesis of various 18F-labelled 3,4-dihydropyrimidin-2-(1H)-ones, imidazo[1,2-a]pyridines, α-acyloxyamides and peptide-type products has been validated. Notably, the chemistry allowed for the 18F-substituent to be positioned on an aryl sub-motif not responsive to direct nucleophilic fluorination. In principle, the technology offers the possibility to label at different positions by using different 18F-components, although preliminary work revealed that all combinations will not necessarily be of comparable efficiency. The practical utility of the methodology was demonstrated in the context of drug discovery with the labelling of the structurally complex target [18F]L-771,668. The methodology is easily implemented and could be adapted to automated systems. Further studies are underway to expand the range of MCRs suitable for 18F-labelling and the pool of radioactive building blocks and to map the various possible combinations of non-labelled and labelled components. The above methodology builds an unprecedented connection between radiochemistry for PET and the field of multicomponent chemistry. Since the development of novel MCRs inclusive of asymmetric transformations is such an active area of research,23 a tantalising range of opportunities emerges for the synthesis of structurally complex 18F-labelled PET tracers but also to access biomarkers for other imaging modalities. This work also establishes that the inclusion of a more convergent retro-radiosynthetic approach in the context of 18F-indirect labelling can greatly expand the range of 18F-labelled radiotracers made accessible for PET.Acknowledgements
This research was supported by the K. C. Wong Education Foundation (L. L.), Hertford College Senior Scholarship (L. L.), the EPSRC (M. H.), GSK (M. H.), the EU (EU-FP7-PEOPLE-IIF-2008-235744) (R. L. Y.) and SIEMENS Molecular Imaging (Project No TP/16636) (R. B.). The authors thank Dr Edward Ball, Dr Jianzhong Zhang, Dr Arkadij Elizarov, Dr Harmuth Kolb at SIEMENS Molecular Imaging for assistance with the microfluidic device.Notes and references
- For selected recent reviews, see (a) M. E. Phelps and J. C. Mazziotta, Science, 1985, 228, 799–801 CAS; (b) H. N. Wagner Jr, J. Nucl. Med., 1991, 32, 561–564; (c) M. E. Phelps, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 9226–9233 CrossRef CAS; (d) Handbook of Radiopharmaceuticals (ed.: M. J. Welch, C. S. Redvanly), Wiley, New York, 2003 Search PubMed; (e) V. J. Cunningham, C. A. Parker, E. A. Rabiner, A. D. Gee and R. N. Gunn, Drug Discovery Today: Technol., 2005, 2, 311–315 CrossRef CAS; (f) PET Chemistry: The Driving Force in Molecular Imaging (ed.: P. A. Schubiger, L. Lehmann, M. Friebee), Springer, New York, 2006 Search PubMed; (g) R. Schirrmacher, C. Wängler and E. Schirrmacher, Mini-Rev. Org. Chem., 2007, 4, 317–329 Search PubMed; (h) S. M. Ametamey, M. Honer and P. A. Schubiger, Chem. Rev., 2008, 108, 1501–1516 CrossRef CAS; (i) D. W. Townsend, J. Nucl. Med., 2008, 49, 938–955 CrossRef; (j) J. M. Hooker, Curr. Opin. Chem. Biol., 2010, 14, 105–111 CrossRef CAS.
- For recent strategies with SPE (Solid-phase Extraction) or F-SPE (Fluorous Solid-Phase Extraction), see: (a) L. J. Brown, D. R. Bouvet, S. Champion, A. M. Gibson, Y. Hu, A. Jackson, I. Khan, N. Ma, N. Millot, H. Wadsworth and R. C. D. Brown, Angew. Chem., Int. Ed., 2007, 46, 941–944 CrossRef CAS; (b) J. W. McIntee, C. Sundararajan, A. C. Donovan, M. S. Kovacs, A. Capretta and J. F. Valliant, J. Org. Chem., 2008, 73, 8236–8243 CrossRef CAS; (c) R. Bejot, T. Fowler, L. Carroll, S. Boldon, J. E. Moore, J. Declerck and V. Gouverneur, Angew. Chem., Int. Ed., 2009, 48, 586–589 CrossRef CAS; (d) E. Blom, F. Karimi and B. Långström, J. Labelled Compd. Radiopharm., 2010, 53, 24–30 CAS.
- P. W. Miller, N. J. Long, R. Vilar and A. D. Gee, Angew. Chem., Int. Ed., 2008, 47, 8998–9033 CrossRef CAS.
- L. Cai, S. Lu and V. W. Pike, Eur. J. Org. Chem., 2008, 2853–2873 CrossRef CAS.
- J. S. Fowler and A. P Wolf, Acc. Chem. Res., 1997, 30, 181–188 CrossRef CAS.
- For selected recent reviews, see: (a) A. Dömling and I. Ugi, Angew. Chem. Int. Ed., 2000, 39, 3168–3210 CrossRef CAS; (b) L. Weber, Drug Discov. Today, 2002, 7, 143–147 CAS; (c) J. Zhu and H. Bienaymé, ed.; Multicomponent Reactions, Wiley–VCH, Weinheim, 2005 Search PubMed; (d) S. D. Bembenek, B. A. Tounge and C. H. Reynolds, Drug Discovery Today, 2009, 14, 278–283 CrossRef CAS.
- For multicomponent reactions based on fluorinated building blocks, see: (a) D. Li, L. Song, S. Song and S. Zhu, J. Fluorine Chem., 2007, 128, 952–957 CrossRef CAS; (b) A. V. Gulevich, N. E. Shevchenko, E. S. Balenkova, G.-V. Röchenthaler and V. G. Nenajdenko, Tetrahedron, 2008, 64, 11706–11712 CrossRef CAS; (c) J. Wu and S. Cao, Curr. Org. Chem., 2009, 13, 1791–1804 CrossRef; (d) H. Yanai and T. Taguchi, Chem. Commun., 2009, 1034–1036 RSC; (e) C. Faggi, A. G. Neo, S. Marcaccini, G. Menchi and Julia Revuelta, Tetrahedron Lett., 2008, 49, 2099–2102 CrossRef CAS.
- (a) V. W. Pike and F. I. Aigbirhio, J. Chem. Soc., Chem. Commun., 1995, 2215–2216 RSC; (b) A. Shah, V. W. Pike and D. A. Widdowson, J. Chem. Soc., Perkin Trans. 1, 1998, 2043–2046 RSC; (c) T. L. Ross, J. Ermert, C. Hocke and H. H. Coenen, J. Am. Chem. Soc., 2007, 129, 8018–8025 CrossRef CAS; (d) J.-H. Chun, S. Lu, Y.-S. Lee and V. W. Pike, J. Org. Chem., 2010, 75, 3332–3338 CrossRef CAS; (e) B. Wang, L. Qin, K. D. Neumannn, S. Uppaluri, R. L. Cerny and S. DiMagno, Org. Lett., 2010, 12, 3352–3355 CrossRef CAS.
- A. Dömling, Chem. Rev., 2006, 106, 17–89 CrossRef.
- (a) G. Vaidyanathan and M. R. Zalutsky, Nat. Protoc., 2006, 1, 1655–1661 CrossRef CAS; (b) J. Ermert and H. H. Coenen, Curr. Radiopharm., 2010, 3, 127–160 Search PubMed; (c) A. Speranza, G. Ortosecco, E. Castaldi, A. Nardelli, L. Pace and M. Salvatore, Appl. Radiat. Isot., 2009, 67, 1664–1669 CrossRef CAS.
- (a) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef; (b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC; (c) C. Faggi, A. G. Neo, S. Marcaccini, G. Menchi and J. Revuelta, Tetrahedron Lett., 2008, 49, 2099–2102 CrossRef CAS.
- C. O. Kappe, Acc. Chem. Res., 2000, 33, 879–888 CrossRef.
- C. O. Kappe, Eur. J. Med. Chem., 2000, 35, 1043–1052 CrossRef CAS.
- (a) K. Groebke, L. Weber and F. Mehlin, Synlett, 1998, 661–663 CAS; (b) H. Bienaymé and K. Bouzid, Angew. Chem., Int. Ed., 1998, 37, 2234–2237 CrossRef CAS; (c) C. Blackburn, B. Guan, P. Fleming, K. Shiosaki and S. Tsai, Tetrahedron Lett., 1998, 39, 3635–3638 CrossRef CAS.
- A. R. Katritzky, Y.–J. Xu and H. Tu, J. Org. Chem., 2003, 68, 4935–4937 CrossRef CAS.
- M. C. Pirrung and K. Das Sarma, J. Am. Chem. Soc., 2004, 126, 444–445 CrossRef CAS.
- (a) C.-C. Lee, G. Sui, A. Elizarov, C. J. Shu, Y.-S. Shin, A. N. Dooley, J. Huang, A. Daridon, P. Wyatt, D. Stout, H. C. Kolb, O. N. Witte, N. Satyamurthy, J. R. Heath, M. E. Phelps, S. R. Quake and H.-R. Tseng, Science, 2005, 310, 1793–1796 CrossRef CAS; (b) A. M. Elizarov, C. E. Ball, J. Zhang, H. Kolb, R. M. van Dam, L. T. Diener, S. Ford and R. Miraghaie, US Patent. 20090036668, 2009; (c) R. Bejot, A. M. Elizarov, C. Ball, J. Zhang, R. Miraghaie, H. C. Kolb and V. Gouverneur, J. Label Compd. Radiopharm, 2010 Search PubMed in press.
- W. C. Eckelman, M. Bonardi and W. Volkert, Nucl. Med. Biol., 2008, 35, 523–527 CrossRef CAS.
- S. Marcaccini and T. Torroba, Nat. Protoc., 2007, 2, 632–639 Search PubMed.
- For selected recent reviews, see (a) M. T. Klimas, Mol. Imaging Biol., 2002, 4, 311–337 Search PubMed; (b) M. Rudin and R. Weissleder, Nat. Rev. Drug Discovery, 2003, 2, 123–131 CrossRef CAS; (c) W. C. Eckelman, Drug Discovery Today, 2003, 8, 404–410 CrossRef CAS; (d) J. W. Dalley, T. D. Fryer, F. I. Aigbirhio, L. Brichard, H. K. Richards, Y. T. Hong, J.-C. Baron, B. J. Everitt and T. W. Robbins, Neuropharmacology, 2009, 56, 9–17 CrossRef CAS.
- Since 18F-fluorine-carbon bond formation is not involved in the MCR step, the specific activity of [18F]2a–5d should be comparable to the one determined for the 18F-building blocks.
- T. G. Murali Dhar, D. Nagarathnam, M. R. Marzabadi, B. Lagu, W. C. Wong, G. Chiu, S. Tyagarajan, S. W. Miao, F. Zhang, W. Sun, D. Tian, Q. Shen, J. Zhang, J. M. Wetzel, C. Forray, R. S. L. Chang, T. P. Broten, T. W. Schorn, T. B. Chen, S. O'Malley, R. Ransom, K. Schneck, R. Bendesky, C. M. Harrell, K. P. Vyas, K. Zhang, J. Gilbert, D. J. Pettibone, M. A. Patane, M. G. Bock, R. M. Freidinger and C. Gluchowski, J. Med. Chem., 1999, 42, 4778–4793 CrossRef CAS.
- In the context of PET, common practice for the production of enantiopure 18F-radiotracers relies on separation of the racemic mixture by chiral stationary phase HPLC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data. NMR Spectra for all novel compounds. Tables of optimization for radio-MCR. Radiolabelling procedures, analysis information and HPLC chromatograms for all 18F-labelled compounds. Specific activity calculations for [18F]1a and [18F]1c. See DOI: 10.1039/c0sc00362j |
| ‡ This publication is part of the web themed issue on fluorine chemistry. |
| § Present address: Singapore Bioimaging Consortium, Agency for Science, Technology and Research (A*STAR), #02-02 Helios, 11 Biopolis Way, Singapore 138667 |
| This journal is © The Royal Society of Chemistry 2011 |