Reactivity modulation in container molecules
Boris
Breiner
,
Jack K.
Clegg
and
Jonathan R.
Nitschke
*
Department of Chemistry, University of Cambridge, Cambridge, CB2 1EW, UK. E-mail: jrn34@cam.ac.uk; Fax: +44(0)1223 336362; Tel: +44(0)1223 336324
First published on 16th August 2010
Abstract
The encapsulation of a guest within a host molecule can open new pathways of guest reactivity or halt modes of reaction normally observed in solution. Recent findings illustrate how a host's size and shape may alter the regioselectivity and kinetics of guests' reactions within host molecules.
 Boris Breiner | Boris Breiner received a Vordiplom in chemistry from the Ruprecht-Karls-Universität in Heidelberg in 2000. He finished his Ph.D. in 2006 after doing research in organic chemistry at the Florida State University, under the supervision of Igor. V. Alabugin and was awarded the Dorothy and Russell Johnsen Dissertation Award. He was subsequently appointed as an instructor at Simmons College and as a postdoctoral associate at the Massachusetts Institute of Technology, in a joint project with Christopher C. Cummins and Daniel G. Nocera. Since 2008, he has been a postdoc in the group of Jonathan R. Nitschke at the University of Cambridge. |
 Jack K. Clegg | Dr Jack K. Clegg completed his undergraduate studies in Chemistry, History and German at the University of Sydney winning The University Medal. He also completed his Bachelor of Laws and PhD at the same institution. His doctoral studies were undertaken under Prof. Leonard F. Lindoy and Prof. Kate A. Jolliffe. Jack has been a Fellow of the Senate of the University of Sydney, a Director of The Australian Youth Orchestra and is the Deputy Chair of The Australian Country Credit Union. He recently won a Marie Curie Fellowship and has taken it up at the University of Cambridge. |
 Jonathan R. Nitschke | Jonathan Nitschke received his bachelor's degree in chemistry from Williams College in 1995 and his doctorate from the University of California, Berkeley in 2001 under the supervision of T. Don Tilley. He then undertook postdoctoral studies with Jean-Marie Lehn in Strasbourg, and in 2003 started his independent research career as a maître-assistant at the University of Geneva. He was the first recipient of the European Young Chemist Award in August 2006 and received the Werner Prize of the Swiss Chemical Society in September 2007. In November 2007 his research group moved to the University of Cambridge. |
Introduction – encapsulation
Numerous biological systems alter the reactivity of one molecule by surrounding it with another in order to achieve a given function. These functions range from the high catalytic activity and substrate selectivity displayed by enzymes, to transport and storage proteins such as transferrin1 and ferritin.2 The former serves as a delivery vehicle for Fe3+ by encapsulating it with high affinity (∼1023 M−1 at physiological pH) and delivering it in a pH-dependent manner inside cells. The latter serves as a storage facility for Fe3+, keeping up to 4500 Fe3+ ions isolated from the rest of the organism, thus protecting it from their toxicity.Using these biological examples for inspiration, comparatively simple artificial systems have been designed in the context of host–guest chemistry3 in order to study the underlying principles of guest encapsulation and the potential for modulation of guest reactivity. Once inside container molecules, guest molecules are confined in tight spaces and isolated from bulk media. Such confinement is reminiscent of the trapping of substrates within biochemical structures – such as enzyme pockets, the interior of proteasomes, or within the ribosome. The behaviour of the isolated guest, whether in an artificial or natural host, diverges substantially from the behaviour of free molecules in solution.4 Predominately, this is due to the random collisions with other molecules that are typically observed in bulk solution being replaced by a smaller set of interactions with the host or a small number of other guest molecules. Confinement within a container molecule can also lead to interesting changes in the physical properties of the trapped guest(s), including inducing unusual spin-states,5 disfavourable conformations6 and exposing the guest(s) to more elevated concentrations and/or pressures than would readily be accessible under ambient conditions.7
Container molecules
A host can physically surround all or part of another molecule. Non-biological host molecules include, but are not limited to, metal–organic cages,8–10 zeolites,11 metal–organic frameworks12 and purely organic capsules4,13 including cavitands,14 cyclodextrins15 and cucurbiturils16 as well as nanotubes17 and fullerenes.18 Container molecules are a subset of host species. To set the boundaries of this minireview we define container molecules as discrete soluble artificial molecules displaying a well-defined inner cavity where the micro-environment of the encapsulated void is altered with respect to the chemical and physical conditions that exist outside the container. In general, it is the presence of this micro-enviroment that leads to the interesting and unusual reactivity of guests within container molecules.Reactivity modulation
As the number of container molecules synthesised increases, so too have the applications of these species. One of the most promising general classes of application is the use of container molecules to alter the reactivity of the species they are hosting. This is of particular interest when the reactivity observed diverges from known behaviour in solution. While container molecules are also useful in other contexts, e.g., gas storage,19 this is not a comprehensive review20 and instead, we highlight recent findings and the most illustrative classic examples of unusual reactivity inside container molecules. To facilitate discussion we define the three following general types of reactivity that have been observed. Firstly, reactions are examined whose courses are altered substantially by the size and shape of the hosts' internal cavity, producing nanoparticles of a particular size, or inducing unusual regioselectivity in substrates. Secondly, container molecules that act as catalysts, lowering activation energies and improving turnover rates of a variety of reactions are explored. Finally, host species that inhibit the reactions of otherwise unstable guest species or allow the isolation of rare reactive intermediates are discussed.Size and shape controlled reactions
A host can influence a reaction within its inner phase simply by providing a cavity of a particular shape and size. This has been shown to be useful in two ways: a well-defined cavity can serve as a mould or endo-template to control the size of a product formed within the cavity, or a tight well-defined binding pocket can lead to preorganisation of substrates and alter the regioselectivity of a given reaction.This approach has found application in the synthesis of uniform silica nanoparticles by employing a container molecule as a template.21 By functionalising the inner surface of a series of self-assembled Pd12L24 spheres with sugar residues, a hydrophilic inner surface suitable for the sol–gel condensation of tetramethoxysilane was created by Fujita et al. (Fig. 1). Depending on the ligand, these spheres had an inner diameter ranging from 2–4 nm and contained 96 inward-facing hydroxyl groups. Upon addition of tetramethoxysilane, sol–gel condensation occurred inside the spherical cavity, yielding silica nanoparticles of particular diameters, relative to dimensions of the sphere employed. The nanoparticles could then be released by the destruction of the sphere. No crosslinking of the nanoparticles or condensation outside the spheres could be detected and the observed polydispersities for these particles approached unity.
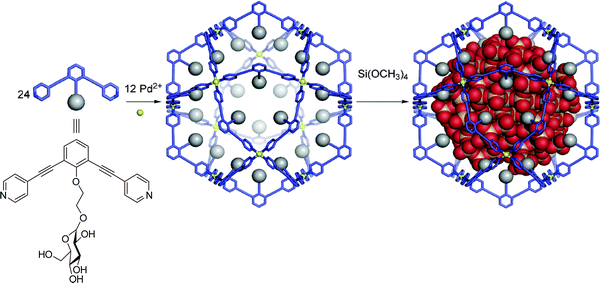 | ||
| Fig. 1 Sol–gel condensation of tetramethoxysilane inside a self-assembled organopalladium sphere produces monodisperse silica nanoparticles. Reproduced with permission.21 | ||
Gibb et al. prepared a water-soluble deep cavitand and encapsulated several methyl cycloalkenes within it. These guest molecules could be photooxidised to allylic hydroperoxides by singlet oxygen. The regiochemistry of the products was controlled in a manner that was previously not possible.22 The formation of the unusual products was enabled by preventing the abstraction of the methyl hydrogens of the substrate. Tight, sterically-hindered binding of the guest within the capsule led to protection of the guests’ methyl groups. The orientation of the methyl cycloalkene was attributed primarily to steric constraints, although a number of C–H⋯π interactions were also present. Similar spatial constraints in the same host species were also shown to be responsible for unexpected products forming from the photodimerisation of 4-methylstyrene.23
Employing a cavitand substituted with four phosphonate groups allowed Dalcanale and Yebeutchou to selectively mono-methylate a variety of primary amines in a procedure that otherwise required tedious purification procedures to separate the desired product from a mixture containing doubly and triply methylated byproducts.24 The selectivity observed is due to strong binding and selective sequestration of the desired product within the host preventing further attack by methyliodide. While the reaction does not take place within the capsule, its size, shape and binding affinity underpin the unusual reactivity.
Diels–Alder reactions between anthracenes and pthalimides generally yield adducts where the central ring (9,10-positions) of the anthracene framework has been bridged as a consequence of the high concentration of π-electron density at those positions. If 9-hydroxymethylanthracene and N-cyclohexylphthalimide, however, are encapsulated within a large self-assembled Pd6L4 octahedron in D2O the reaction yields the 1,4-Diels–Alder product (Fig. 2a).9 Similarly, an otherwise notoriously stubborn Diels–Alder reaction between a substituted napthalene and N-cyclohexylphthalimide was shown to proceed within the same capsule yielding the unexpected syn-1,4-product (Fig. 2b). If instead of heating, the same species were irradiated with a mercury lamp the [2 + 2] photoaddition proceeds to yield the 1,2-substituted product following a reaction path unknown in solution (Fig. 2c).25 The host species facilitates the interesting regio- and stereoselectivity of these reactions by tight packing of the substrates within its central cavity leading to efficient preorganisation with close proximity between the reaction positions and the dienophile and precluding any rearrangements. The stability of the host–guest complex was attributed to a combination of π stacking and the hydrophobic effect.
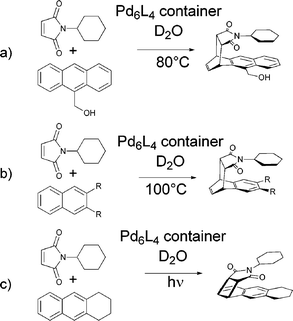 | ||
| Fig. 2 Three cycloaddition reactions yielding products with unusual regio- and stereoselectivity when carried out within an octahedral Pd6L4 container. | ||
Catalytic reactions
Catalysts increase the rate of a given reaction by providing a pathway with reduced activation energy. Enzymatic catalysts do so by stabilising transition states and reactive intermediates while also isolating them, protecting these species from undesired reactions with other molecules.26 A number of container molecules have been shown to function in a similar way, dramatically increasing the rate of reactions taking place within them.Again investigating aqueous Diels–Alder reactions, Fujita's group demonstrated that a bowl-shaped container (rather than the octahedron discussed above) served as a catalyst (Fig. 3).9 In the presence of the Pd6L4 bowl (1 mol%), 9-hydroxymethylanthracene and N-phenylmaleimide reacted to form the expected 9,10-adduct in quantitative yield. In the absence of the catalyst, the reaction barely took place, with a yield of only 3% under otherwise identical conditions. Although the high-energy intermediate species were stabilised by π-stacking in both the octahedral cage and bowl-shaped container, catalysis was observed only in the latter because its binding of the starting materials was far more favourable than product binding. Hence, the product was efficiently ejected from the host following the reaction.
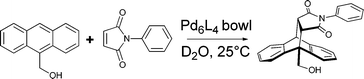 | ||
| Fig. 3 Catalysis of the 9,10-Diels–Alder addition between 9-hydroxymethylanthracene and N-phenylmaleimide within a bowl-shaped container. | ||
Covalently formed organic calix[4]arene derivatives are open on one end and closed at the other, an arrangement similar to the metal–organic bowl just described. One such calixarene, equipped with an inwardly directed carboxylic acid group, has been employed by the group of Rebek to catalyse the cyclisation of an epoxyalcohol.27 The carboxylic acid moiety assists in substrate binding through hydrogen bonding interactions and also activates the epoxyalcohol guest. Internal π-electron density serves to stabilise the intermediates through cation–π interactions. The authors propose a catalytic cycle (Fig. 4) that bears a close resemblance to enzymatic pathways. The kinetics of the systems are Michaelis–Menten-like, where the rate-limiting step is the reaction of a substrate–catalyst complex to yield a product–catalyst complex. A number of reactions between carboxylic acids and isonitriles have been observed to be accelerated inside similar organic cavitand container molecules.28,29
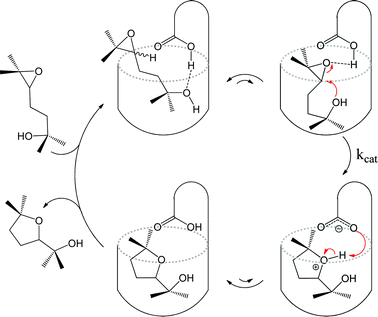 | ||
| Fig. 4 Proposed catalytic cycle for regioselective cyclisation of an epoxyalcohol inside a synthetic cavitand receptor. | ||
Despite the proposed enzyme-like pathway of the reactions employed by both the bowl-like structures or cavitands, the catalytic rate enhancement by these species is significantly slower than the highest accelerations produced by enzymes.
The first example of a container molecule capable of reaction acceleration by a factor of 106 – a degree of rate enhancement that had belonged exclusively to the domain of enzymes – was provided by the groups of Raymond and Bergman. Their tetrahedral M4L6 (M = GaIII) coordination cage was shown to accelerate the Nazarov cyclisation of 1,4-pentadien-3-ols by as much as 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100000-fold, compared to the same reaction in bulk solution.30 The same cage was also observed to catalyse a number of proton mediated reactions,31 including acetal hydrolysis (Fig. 5)32 and aza-Cope rearrangements.33 This is remarkable because these acid-catalysed reactions could be performed despite the surrounding bulk solution being basic. The anionic, π-electron-rich cage structure served to stabilise the cationic intermediate of acetal hydrolysis.
100000-fold, compared to the same reaction in bulk solution.30 The same cage was also observed to catalyse a number of proton mediated reactions,31 including acetal hydrolysis (Fig. 5)32 and aza-Cope rearrangements.33 This is remarkable because these acid-catalysed reactions could be performed despite the surrounding bulk solution being basic. The anionic, π-electron-rich cage structure served to stabilise the cationic intermediate of acetal hydrolysis.
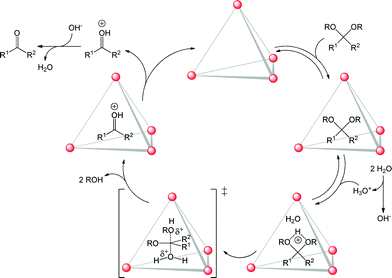 | ||
| Fig. 5 Proposed catalytic cycle for acid-catalysed acetal deprotection inside a self-assembled tetrahedral host in basic media. | ||
In order to catalyse its guest's transformation, a host must strike a balance between enclosing a space well enough to differentiate it from the bulk environment, while remaining flexible enough to allow for reactant uptake and product liberation, as exemplified by the system of Fig. 5.
Inhibition of reactivity
In the same way that binding of guest molecules within container molecules can stabilise unusual intermediates, leading to interesting regio- and stereoselectivity or catalytic behaviour, otherwise highly unstable molecules or intermediates can be isolated, identified and stored through encapsulation. In a classic example, Cram et al. employed a covalently constructed carcerand to stabilise cyclobutadiene (Fig. 6).34 Irradiating α-pyrone trapped within the cavity leads to a chain of chemical reactions inside the container yielding cyclobutadiene and carbon dioxide. The latter is expelled through the pores of the capsule, leaving cyclobutadiene within the host. Thus protected, cyclobutadiene could be characterised by NMR spectroscopy for the first time. Using similar containers, Warmuth succeeded in isolating and characterising o-benzyne35 and Rebek and Restorp observed isoimides of great hydrolytic sensitivity.29 Fujita's group has stabilised cyclotrisiloxanes in a self-assembled sphere36 and succeeded in directly observing a coordinatively unsaturated molybdenum-methylcyclopentadienyl species by crystallography.37![Isolation of cyclobutadiene within a carcerand derived from calix[4]arene.](/image/article/2011/SC/c0sc00329h/c0sc00329h-f6.gif) | ||
| Fig. 6 Isolation of cyclobutadiene within a carcerand derived from calix[4]arene. | ||
White phosphorous, tetrahedral P4, is violently pyrophoric, spontaneously combusting upon exposure to atmospheric dioxygen. Our group recently demonstrated that a self-assembled, water-soluble, tetrahedral iron-containing container molecule encapsulates P4, thus rendering it water-soluble and air-stable.38 Since the pores of the cage (Fig. 7) are large enough to allow oxygen to pass through,10 stabilisation of reactive P4 is not achieved by excluding oxygen, but rather by constriction of the guest. As the cage is too small to accommodate the intermediates of the oxidation reaction, the highly exothermic reaction is prevented.
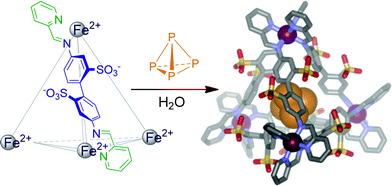 | ||
| Fig. 7 Encapsulation of white phosphorus by a self-assembled tetrahedral cage complex. | ||
Future directions
One potential application of container molecules' reactivity modulation lies in the context of drug delivery. A biologically active compound could be protected by a surrounding host molecule and carried to a specific location in an organism, where, upon application of a particular stimulus, the encapsulated species would be released. This would concentrate the therapeutic effect in a targeted region and hence minimise problems such as unwanted drug side effects, general toxicity and poor solubility. Rapid progress in this direction is possible as container molecules that disassemble/reassemble upon application of an outside stimulus (e.g., a change in pH) already exist.10,39Catalytic reactions will also remain a topic of interest, with new routes to tremendous rate enhancements and high selectivity of significant interest to industry. A wealth of new reaction pathways may be screened and optimised using the tools of dynamic combinatorial chemistry,40 which could enable the creation of host–guest systems capable of adaptive behaviour – altering their size and the shape of their inner void by reacting with a library of small molecules of complementary functionality.
Finally, the use of hosts as “whole molecule protecting groups” for specific guests is of interest in the context of systems chemistry,41 whereby the reactivity of an encapsulated species may be turned on upon application of a stimulus, by liberating it from a host.
Conclusions
The use of host molecules to shape guest reactivity provides an excellent example of how the application of fundamental principles, originally discovered in biological contexts, can result in new and useful chemistry. The design and discovery of new host architectures will allow for new regio- and stereoselective catalytic transformations as the whole space around a substrate, not just the reactive centre, is controlled. The years to come promise rapid advances as principles of host design are discovered and codified, and as new applications beckon with the discovery of each new host structure.42Notes and references
- A. Dautry-Varsat, A. Ciechanover and H. F. Lodish, Proc. Natl. Acad. Sci. U. S. A., 1983, 80, 2258–2262 CAS.
- E. C. Theil, Annu. Rev. Biochem., 1987, 56, 289–315 CrossRef CAS.
- D. J. Cram and J. M. Cram, Science, 1974, 183, 803–809 CrossRef CAS.
- J. Rebek, Acc. Chem. Res., 2009, 42, 1660–1668 CrossRef.
- K. Ono, M. Yoshizawa, M. Akita, T. Kato, Y. Tsunobuchi, S. Ohkoshi and M. Fujita, J. Am. Chem. Soc., 2009, 131, 2782–2783 CrossRef CAS.
- D. Ajami and J. Rebek, Jr., Nat. Chem., 2009, 1, 87–90 CrossRef CAS; M. R. Ams, D. Ajami, S. L. Craig, J.-S. Yang and J. Rebek Jr., J. Am. Chem. Soc., 2009, 131, 13190–13191 CrossRef CAS.
- M. Saunders, Helv. Chim. Acta, 2003, 86, 1001–1007 CrossRef CAS; D. Ajami and J. Rebek, Jr., Angew. Chem., Int. Ed., 2008, 47, 6059–6061 CAS.
- R. W. Saalfrank, A. Stark, K. Peters and H. G. Von Schnering, Angew. Chem., 1988, 100, 878–880 CrossRef CAS; M. D. Ward, Chem. Commun., 2009, 4487–4499 RSC; R. W. Saalfrank, H. Maid and A. Scheurer, Angew. Chem., Int. Ed., 2008, 47, 8794–8824 CrossRef CAS; C. R. K. Glasson, G. V. Meehan, J. K. Clegg, L. F. Lindoy, P. Turner, M. B. Duriska and R. Willis, Chem. Commun., 2008, 1190–1192 RSC; D. J. Tranchemontagne, Z. Ni, M. O'Keeffe and O. M. Yaghi, Angew. Chem., Int. Ed., 2008, 47, 5136–5147 CrossRef CAS; M. Albrecht and R. Froehlich, Bull. Chem. Soc. Jpn., 2007, 80, 797–808 CrossRef CAS; M. A. Pitt and D. W. Johnson, Chem. Soc. Rev., 2007, 36, 1441–1453 RSC; M. J. Hardie, Chem. Soc. Rev., 39, pp. 516–527 Search PubMed; J. K. Clegg, L. F. Lindoy, B. Moubaraki, K. S. Murray and J. C. McMurtrie, Dalton Trans., 2004, 2417–2423 Search PubMed; D. L. Caulder and K. N. Raymond, Acc. Chem. Res., 1999, 32, 975–982 RSC; P. Mal and J. R. Nitschke, Chem. Commun., 2010, 46, 2417–2419 CrossRef CAS.
- M. Yoshizawa, M. Tamura and M. Fujita, Science, 2006, 312, 251–254 CrossRef CAS.
- P. Mal, D. Schultz, K. Beyeh, K. Rissanen and J. R. Nitschke, Angew. Chem., Int. Ed., 2008, 47, 8297–8301 CrossRef CAS.
- T. Bein, Stud. Surf. Sci. Catal., 2007, 168, 611–657 CAS.
- R. P. John, D. Moon and M. S. Lah, Supramol. Chem., 2007, 19, 295–308 CrossRef CAS; B. Breiner and J. R. Nitschke, Nat. Chem., 2010, 2, 6–7 CrossRef CAS; J. K. Clegg, S. S. Iremonger, M. J. Hayter, P. D. Southon, R. B. MacQuart, M. B. Duriska, P. Jensen, P. Turner, K. A. Jolliffe, C. J. Kepert, G. V. Meehan and L. F. Lindoy, Angew. Chem., Int. Ed., 2010, 49, 1075–1078 CrossRef CAS; G. J. Halder, C. J. Kepert, B. Moubaraki, K. S. Murray and J. D. Cashion, Science, 2002, 298, 1762–1765 CrossRef CAS.
- S. J. Dalgarno, J. L. Atwood and C. L. Raston, Chem. Commun., 2006, 4567–4574 RSC; S. Liu and B. C. Gibb, Chem. Commun., 2008, 3709–3716 RSC; E. Huerta, G. A. Metselaar, A. Fragoso, E. Santos, C. Bo and J. de Mendoza, Angew. Chem., Int. Ed., 2007, 46, 202–205 CrossRef CAS.
- W. Sliwa and J. Peszke, Mini-Rev. Org. Chem., 2007, 4, 125–142 Search PubMed.
- R. Breslow and S. D. Dong, Chem. Rev., 1998, 98, 1997–2011 CrossRef CAS.
- L. Isaacs, Chem. Commun., 2009, 619–629 RSC.
- V. G. Organo and D. M. Rudkevich, Chem. Commun., 2007, 3891–3899 RSC; S.-Y. Hong, R. Popovitz-Biro, G. Tobias, B. Ballesteros, B. G. Davis, M. L. H. Green and R. Tenne, Nano Res., 2010, 3, 170–173 CrossRef CAS.
- M. Takata, B. Umeda, E. Nishibori, M. Sakata, Y. Saito, M. Ohno and H. Shinohara, Nature, 1995, 377, 46–49 CrossRef CAS; D. S. Bethune, R. D. Johnson, J. R. Salem, M. S. de Vries and C. S. Yannoni, Nature, 1993, 366, 123–128 CrossRef CAS.
- S. J. Dalgarno, P. K. Thallapally, L. J. Barbour and J. L. Atwood, Chem. Soc. Rev., 2007, 36, 236–245 RSC; M. B. Duriska, S. M. Neville, J. Lu, S. S. Iremonger, J. F. Boas, C. J. Kepert and S. R. Batten, Angew. Chem., Int. Ed., 2009, 48, 8919–8922 CrossRef CAS; C. Pariya, C. R. Sparrow, C.-K. Back, G. Sandi, F. R. Fronczek and A. W. Maverick, Angew. Chem., Int. Ed., 2007, 46, 6305–6308 CrossRef CAS.
- M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS.
- K. Suzuki, S. Sato and M. Fujita, Nat. Chem., 2010, 2, 25–29 CrossRef CAS.
- A. Natarajan, L. S. Kaanumalle, S. Jockusch, C. L. D. Gibb, B. C. Gibb, N. J. Turro and V. Ramamurthy, J. Am. Chem. Soc., 2007, 129, 4132–4133 CrossRef CAS.
- A. Parthasarathy, L. S. Kaanumalle and V. Ramamurthy, Org. Lett., 2007, 9, 5059–5062 CrossRef CAS.
- R. M. Yebeutchou and E. Dalcanale, J. Am. Chem. Soc., 2009, 131, 2452–2453 CrossRef CAS.
- T. Murase, S. Horiuchi and M. Fujita, J. Am. Chem. Soc., 2010, 132, 2866–2867 CrossRef CAS.
- D. Ringe and G. A. Petsko, Science, 2008, 320, 1428–1429 CrossRef CAS; R. J. Hooley and J. Rebek, Chem. Biol., 2009, 16, 255–264 CrossRef CAS.
- F. R. Pinacho Crisostomo, A. Lledo, S. R. Shenoy, T. Iwasawa and J. Rebek, Jr., J. Am. Chem. Soc., 2009, 131, 7402–7410 CrossRef CAS.
- J.-L. Hou, D. Ajami and J. Rebek, Jr., J. Am. Chem. Soc., 2008, 130, 7810–7811 CrossRef CAS.
- P. Restorp and J. Rebek, Jr., J. Am. Chem. Soc., 2008, 130, 11850–11851 CrossRef CAS.
- C. J. Hastings, M. D. Pluth, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2010, 132, 6938–6940 CrossRef CAS.
- M. D. Pluth, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2009, 42, 1650–1659 CrossRef CAS.
- M. D. Pluth, R. G. Bergman and K. N. Raymond, Science, 2007, 316, 85–88 CrossRef CAS.
- C. J. Hastings, D. Fiedler, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2008, 130, 10977–10983 CrossRef CAS.
- D. J. Cram, M. E. Tanner and R. Thomas, Angew. Chem., Int. Ed., 1991, 30, 1024–1027 CrossRef.
- R. Warmuth, Angew. Chem., Int. Ed. Engl., 1997, 36, 1347–1350 CrossRef CAS.
- M. Yoshizawa, T. Kusukawa, M. Fujita and K. Yamaguchi, J. Am. Chem. Soc., 2000, 122, 6311–6312 CrossRef CAS.
- M. Kawano, Y. Kobayashi, T. Ozeki and M. Fujita, J. Am. Chem. Soc., 2006, 128, 6558–6559 CrossRef CAS.
- P. Mal, B. Breiner, K. Rissanen and J. R. Nitschke, Science, 2009, 324, 1697–1699 CrossRef CAS.
- D. J. Bray, B. Antonioli, J. K. Clegg, K. Gloe, K. Gloe, K. A. Jolliffe, L. F. Lindoy, G. Wei and M. Wenzel, Dalton Trans., 2008, 1683–1685 RSC.
- P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J. L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS; J. M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC; S. Otto and S. Kubik, J. Am. Chem. Soc., 2003, 125, 7804–7805 CrossRef CAS.
- J. R. Nitschke, Nature, 2009, 462, 736–738 CrossRef CAS; R. F. Ludlow and S. Otto, Chem. Soc. Rev., 2008, 37, 101–108 RSC.
- Q.-F. Sun, J. Iwasa, D. Ogawa, Y. Ishido, S. Sato, T. Ozeki, Y. Sei, K. Yamaguchi and M. Fujita, Science, 2010, 328, 1144–1147 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |