Unravelling the fine structure of stacked bipyridine diamine-derived C3-discotics as determined by X-ray diffraction, quantum-chemical calculations, Fast-MAS NMR and CD spectroscopy†
Thorsten
Metzroth
a,
Anke
Hoffmann
b,
Rafael
Martín-Rapún
c,
Maarten M. J.
Smulders
c,
Koen
Pieterse
c,
Anja R. A.
Palmans
c,
Jef A. J. M.
Vekemans
c,
E. W.
Meijer
*c,
Hans W.
Spiess
*b and
Jürgen
Gauss
*a
aInstitut für Physikalische Chemie, Universität Mainz, D-55099, Mainz, Germany. E-mail: gauss@uni-mainz.de
bMax-Planck-Institute for Polymer Research, Postfach 3148, D-55021, Mainz, Germany. E-mail: spiess@mpip-mainz.mpg.de
cLaboratory of Macromolecular and Organic Chemistry, Eindhoven University of Technology, P.O. Box 513, 5600 MB, Eindhoven, The Netherlands. E-mail: e.w.meijer@tue.nl
First published on 9th September 2010
Abstract
An in depth investigation of the fine structure adopted by the helical stacks of C3-discotics 1 incorporating three 3,3′-diamino-2,2′-bipyridine units is described. In the bulk the molecules display liquid crystalline behaviour in a temperature window of >300 K and an ordered rectangular columnar mesophase (Colro) with an inter-disc distance of 3.4 Å is assigned. X-Ray diffraction on aligned samples has also revealed a helical superstructure in the liquid crystalline state, and a rotation angle of 13–16° between consecutive discs. The proposed superstructure in the bulk phase has been further substantiated by a combination of quantum-chemical calculations and solid-state NMR spectroscopy. Dilute solution NMR spectroscopy and elaborate CD spectroscopy on aggregated samples have revealed an isodesmic growth pattern of the C3-discotics. From the combined results it has become evident that the fine tuning interaction responsible for the highly ordered helical architectures is not weak intermolecular hydrogen bonding, but rather rigidification, due to propeller formation after preorganisation by π–π interactions. Although all the techniques used underpin the structural features proposed, none of them individually is able to point to a unique structure. However, together the techniques give very strong evidence for a confined ship-screw arrangement in which all amidic carbonyl oxygens point in one direction.
Introduction
The helix motif is ubiquitously found in Nature and intrinsically linked to living matter. Helical superstructures in biomolecules, such as the double helix in DNA or the α-helix in proteins, impart crucial functions such as recognition, replication and selective activity. Also in modern life, helical structures are of prominent interest. In the field of chirality sensors, responsive materials and chiroptical devices, molecules that order in a helical fashion –macromolecularly or supramolecularly – have attained a great deal of attention.1,2 In fact, cholesteric liquid crystals –showing a helical order between the molecules in the mesophase– are one of the few organic materials frequently applied in electronic devices.3Liquid crystals usually consist of a rigid core and flexible side chains, and the development of large extended aromatic cores resulted in discotic liquid crystals with increased mesophase stability.4–10 Discotic liquid crystals based on the 3,3′-diamino-2,2′-bipyridine unit (compound 1, Fig. 1) showed exceptionally stable mesophases of >300 K11 and self-assembled C3-discotics 1a–c, carrying hydrophobic peripheral mesogenic gallic moieties, display highly ordered helical structures.12X-Ray diffraction on the liquid-crystal phases proved the tendency to form ordered columnar mesophases11 while in dilute solution, in non-solvents such as dodecane, transfer of peripheral chirality, present in the individual molecules, to the central part of the aggregated structure led to the expression of the “Sergeant-and-soldiers principle”12 and “Majority-rules”.13
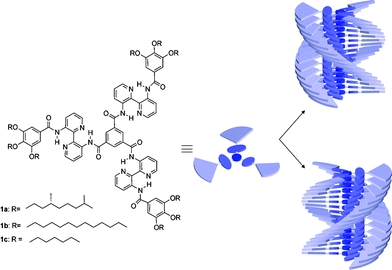 | ||
| Fig. 1 Investigated molecules 1a–c and their originally proposed helical superstructure. In chiral 1a one helical sense is preferred, while for 1b,c both helical senses are present in equal amounts. | ||
The remarkable properties of these C3-discotics were ascribed to a combination of secondary interactions including strong six-fold intramolecular hydrogen bonding, phase separation of the center and periphery, considerable π–π interactions due to benzene and pyridine nuclei and – last but not least – fine-tuned three-fold intermolecular hydrogen bonding between the central amide functionalities reminiscent of the α-helix formation in peptides.12 The latter interpretation was difficult to prove in view of the weakness of the interaction but we believed that our view was strongly supported by the appearance of a single-crystal structure of the much smaller N,N′,N′′-tris(2-methoxyethyl)benzene-1,3,5-tricarboxamide, displaying a three-fold array of intermolecular hydrogen bonding with a tilt of the amide functions by 40° with respect to the π–π stacked benzene cores, and a rotation of superimposed molecules by 60° to accommodate every molecule in an infinite crystal.14,15 Moreover, it turned out that analogous chiral N,N′,N′-tris(dihydrocitronellyl)benzene-1,3,5-tricarboxamide was subject to comparably strong “Sergeant-and-soldiers principle”16 and “Majority-rules”17–19 behaviour. All efforts to prove this view with the aid of a single-crystal structure of a diaminobipyridineC3-discotic failed, however, either because the analogue did not show a melting point above room temperature (hexyl, dodecyl, dihydrocitronellyl) or because the quality of the crystals could not be optimised sufficiently (methyl, propyl, octadecyl).
In this paper, we describe our efforts to further unravel the fine structure of stacks of representative 3,3′-diamino-2,2′-bipyridine-derived C3-discotics, chiral 1a and achiral 1b and 1c, (Fig. 1)11 and to further improve our understanding of the supramolecular interactions underlying the creation of highly ordered helical superstructures in the liquid crystalline phase and in dilute solution. To elucidate more of the fine structure of these C3-discotics complementary methods were applied, namely X-ray diffraction, quantum-chemical calculations, solid- and solution-state NMR and CD spectroscopy. The reason for such a combined approach is intuitively clear: while a helical packing can be observed by X-ray diffraction and CD spectroscopy, the structural conformation of the cores in the stack remains unsolved. Insight into this question can be gained by a combination of NMR experiments and quantum-chemical calculations where the latter allows for finding a model that can be used to assign the measured NMR spectra.
Results and discussion
X-Ray diffraction of 1a,b in the liquid crystalline phase
The bipyridinyl-based molecules under study exhibit liquid crystalline behaviour from below RT to above 300 °C, when decomposition starts.11 According to polarised optical microscopy observations the mesophase is columnar. In order to gather information on the intra- and intercolumnar organisation in the liquid crystalline phase, we have performed temperature-dependent X-ray diffraction experiments on macroscopically aligned samples of compounds 1a and 1b. The alignment was achieved by shearing the sample inside a capillary at the temperature of the mesophase at which the material is sufficiently fluid (∼170 °C). The X-ray patterns are qualitatively the same for both 1a and 1b and are consistent with those previously reported.11In Fig. 2a we show the wide angle X-ray diffraction pattern for 1b. An amplification of the small angle region is shown in ESI† (Fig. S2). In the wide angle region the pattern exhibits the diffuse halo corresponding to the disordered aliphatic chains and two maxima centered on the meridian. The outer one corresponds to an inter-disc distance of 3.4 Å along the columns. This value indicates that the aromatic cores of the discs are perpendicular to the columnar axis. The slightly weaker, inner refection at 3.7 Å could not be indexed unambiguously but, as discussed below, is indicative of an additional structural detail beyond the simple intermolecular distance between consecutive molecules in one column. In the small-angle region, the most intense maximum lies on the equator (Fig. 2b) and is associated only to the 2D order in the plane perpendicular to the columnar axis (plane XY). However, at least five other maxima in this region exhibit a four-point pattern (Fig. 2b). All these Bragg reflections can be indexed with a space lattice defined by three orthogonal vectors a, b and c (see ESI†, Table S1). Vectors a and b are related to the two-dimensional rectangular lattice (P21 symmetry) in the plane XY perpendicular to the axis of the columns whereas c is parallel to that axis. Interestingly, all the reflections showing the four-spot pattern are located in a layer with the same Miller index l = 1. The fact that c is needed to define the space lattice means that there is a modulation in the electron density along the columns and that there is a correlation between neighbouring columns.
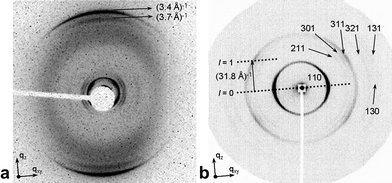 | ||
| Fig. 2 X-Ray diffraction patterns of aligned samples of 1b. The shearing direction and axis of the columns coincide with the direction of qz, whereas plane XY is perpendicular to the axis of the columns. (a) Wide-angle X-ray diffraction pattern of 1b at 120 °C obtained with synchrotron radiation (an amplification of the small angle region is available in ESI†, Fig. S2 and S3). (b) Small-angle X-ray diffraction pattern of 1b at 120 °C. The periodicity of the helix (p) equals the modulus of c (c = 31.8 Å) and corresponds to a 120°-turn (360°/3) of the helix under the C3 symmetry of 1b. | ||
Given the results from other techniques that show that 1a,b and analogous compounds self-assemble in solution in a helical manner,12 it is straightforward to ascribe the modulation of electron density to the columns being helical. Furthermore, similar X-ray patterns have been previously described for other materials displaying a columnar mesophase with helical columns, such as natural20 and artificial polymers,21 metallomesogens,22 discotics23,24 and dendritic25 or star-shaped molecules.26 The periodicity (p) along the axis of the columns must equal the modulus of c (Fig. 2b), that is, 31.8 Å in the case of 1b, and must correspond to a 120°-turn of the helix given the C3-symmetry of the molecule. If taking into account the π–π stacking distance of 3.4 Å between individual molecules, a non-integer number of molecules, 9.35 (about 28 molecules for a 360°-turn), would be needed for the 120°-turn of the helix. As the number of molecules per turn is not an integer, there is no translational periodicity along the direction of the columns and c is not truly a lattice parameter. Additional reflections should theoretically appear with different values of l,27 however none of these reflections are detected in our X-ray patterns. Since about nine molecules are needed for a 120°-turn of the helix, one molecule is rotated approximately 13° with respect to its immediate neighbours in the same stack.
A similar analysis is feasible for 1a, which is enantiomerically pure. Miller indices change but there is a similar rotation φ about 16° between consecutive monomers. In Table 1 we gather the main parameters for the helical structures of both compounds. Indexing of all reflections according to an orthorhombic lattice with P21 symmetry in the XY-plane and with a periodicity p along the column axis is available in the ESI† for both compounds.
| 1a | 1b | |
|---|---|---|
| Pitch (120°)/Å | 25.5 | 31.8 |
| Inter-disc distance/Å | 3.38 | 3.40 |
| Number of discs for a 120°-turn | 7.54 | 9.35 |
| Rotation angle φ between consecutive discs | 16° | 13° |
The most important parameter describing the helix is φ or the rotation angle between consecutive discs. Angle φ derives from the combination of inter-disc distance and helical pitch. The inter-disc distance is approximately the same for both compounds. This seems counterintuitive, but the branching can have a minor effect on the distances along the columns, as long as there is a propeller-like conformation and a helicoidal arrangement. Thus the branching would hinder the stacking between the gallic moieties in a helicoidal structure as that shown in Fig. 1 and would contribute to a larger twist of the helix, and as a result, to a decrease of the helical pitch. In a propeller-like conformation of the molecules within the stack, the maximum with reciprocal spacing 3.7 Å, could correspond to the distance between the centroids of the bipyridine moieties. It is remarkable that φ is only about 16°, far remote from that observed in single crystals of smaller N,N′,N′′-trialkylbenzene-1,3,5-tricarboxamides which is 60°.14,15 This fact would exclude for the bipyridine-based discs the same kind of intermolecular hydrogen bonding which is the driving force for the formation of stacks of the smaller NN′,N′-trialkylbenzene-1,3,5-tricarboxamides. To conclude, an ordered columnar rectangular mesophase (Colro) in which the columns are inherently helical, is the most plausible structure in view of our diffraction patterns. The absence of reflections at different l values, as expected from incommensurate helices, makes this assignment not conclusive though. Therefore, results from other techniques as well as calculations are required to provide further support for the proposed structure.
Calculations
Quantum-chemical calculations were performed to model the dependence of the NMR spectra on the structural arrangement of 1 in the solid state. As the peripheral gallic moieties are of minor relevance for the interpretation of the experimental 1H and 13C NMR spectra, calculations were carried out for the simplified C3-symmetrical system 2 as well as corresponding oligomers. For the monomer, calculations were performed using density-functional theory (DFT)28 and second-order Møller–Plesset perturbation theory (MP2),29 together with a polarized split-valence (SVP) basis set30 (see ESI† for a full documentation of all computational details). Both the DFT and MP2 calculations yield an equilibrium geometry for the monomer with the three bipyridinyl substituents slightly twisted out of the aromatic core plane (see Fig. 3). The obtained distances and angles agree reasonably well between the two sets of calculations with the largest deviation seen for the bipyridinyl torsion angle. There, the angle obtained at the DFT level (3.2°) differs by about 10° from the corresponding MP2 value (12.9°). The differences for the bond angles and bond distances are much smaller (i.e., at most a few degrees and a few pm, respectively), thus indicating the overall reliability of the obtained structure. The relevant tilt angle of the amide with respect to the central benzene unit amounts to 6.7 and 12.5°, applying DFT and MP2 calculations, respectively.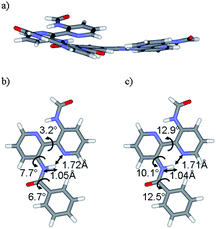 | ||
| Fig. 3 (a) Structure of model 2 as obtained in the quantum-chemical calculations at RIDFT-BLYP/SVP and RI-MP2/SVP level. The detailed structure of a side chain is shown in (b) (DFT results) and (c) (MP2 results) together with a selected set of bond distances (in Å) and bond angles (in degrees). | ||
To obtain a structural model for the superstructure of model 2 in the stack, the optimal rotation angle φ between two superimposed monomers has been determined in calculations for a dimeric model as depicted in Fig. 4. It has to be stressed that we do not and cannot aim at an accurate description of the rotation angle. Rather, the existence of angle dependence allows for a theoretical model of the stack. In conjunction with NMR shift calculations, such a crude model can provide information for the assignment of experimental NMR spectra. The functional dependence of the energy on the rotation angle is shown in Fig. 4. The energetic minimum is obtained around 28°, but given the flatness of the energy curve in conjunction with the discussed inevitable crudeness of the model, angles ranging from 10 to 40° have to be taken into consideration.
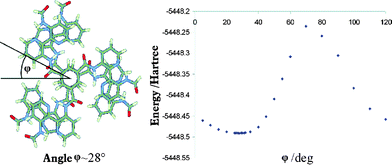 | ||
| Fig. 4 Definition of the rotation angle φ shown for a dimer of model 2 and functional dependence of the dimer energy on the angle φ as obtained in RI-DFT/SVP calculations. | ||
Deviations in chemical shifts are small (at most 0.3 ppm for 1H and 2 ppm for 13C chemical shifts) (Table 2), indicating that the rotation angle does not strongly affect chemical shieldings of the investigated compound. This being the case, we selected one geometry (φ = 28°) for the calculations on the pentamer, for reasons of computational costs.
| Proton | 1-mer 2 | 3-mer 2 | 5-mer 2 | Solid 1b | Solution 1c | |
|---|---|---|---|---|---|---|
| 28° | 13° | |||||
| Hortho | 9.5 | 8.5 | 8.5 | 7.6 | 6.7 | 9.15 |
| H4 | 10.1 | 9.7 | 9.8 | 9.0 | 7.3 | 9.54 |
| H5 | 7.1 | 6.7 | 6.5 | 6.4 | 5.7 | 7.50 |
| H6 | 7.9 | 7.6 | 7.3 | 7.4 | 6.7 | 8.38 |
| H4′ | 9.7 | 9.7 | 9.4 | 9.5 | 8.7 | 9.38 |
| H5′ | 7.2 | 7.0 | 6.9 | 6.5 | 6.0 | 7.45 |
| H6′ | 9.3 | 8.6 | 8.7 | 7.9 | 7.8 | 9.02 |
| NHinner | 16.5 | 16.0 | 15.9 | 15.3 | 13.8 | 15.47 |
| NHouter | 14.6 | 13.8 | 13.8 | 13.7 | 13.0 | 14.36 |
For the protons, the calculations indicate that there are strong neighbouring effects on the shifts due to the π–π stacking. The π shifts induced by the aromatic moieties beneath and above the considered disc lead to high-field shifts in the computed shieldings. These shifts are larger for the pentamer than for the trimer and are expected to be even more pronounced for the heptamer for which a quantum-chemical calculation currently is not feasible. The calculated high-field shifts are about 1.19 ppm (ortho H), 0.83 ppm (inner N–H), and 0.89 ppm (outer N–H) when going from the monomer to the trimer and about 0.9 ppm (ortho H), 0.75 ppm (inner N–H) and 0.13 (outer N–H) when going from the trimer to the pentamer.
It appears that in the model lacking the gallic moieties, convergence of the proton shifts with the stack size is rather slow, and that our computed values for the pentamer only provide upper limits for the actual solid-state upfield proton shifts. This conclusion is in line with results for the 1H NMR shifts of columnar stacks of substituted hexabenzocoronenes where a similarly slow convergence with stack size is observed.28 A faster convergence is reported only in cases where a herringbone structure of hexabenzocoronenes is present.29 The convergence with stack size is also illustrated in the bottom part of Fig. 5 where the calculated 1H NMR spectra for monomer, trimer and pentamer of 2 are compared.
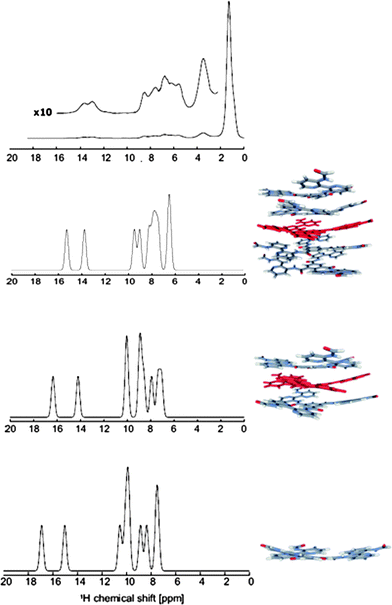 | ||
| Fig. 5 Comparison of the solid-state 1H NMR spectrum of 1b (top) with those derived from the computed NMR chemical shifts for monomer, trimer and pentamer of model 2. | ||
For the 13C NMR chemical shifts (see Table 3), a comparison of the computed values for monomer and trimer indicates that π shifts and, thus, stacking effects are much less pronounced than for the protons. The differences between the computed values for monomer and trimer are in most cases less than 1 ppm, and only for carbons 6 and 6′, about 3 ppm.
| Carbon | Monomer 2 | 3-mer 2 | Solid 1b | Solution 1c | |
|---|---|---|---|---|---|
| 28° | 13° | ||||
| Cortho | 130 | 131 | 132 | 128 | 129.4 |
| C4 | 127 | 126 | 128 | 127 | 129.6 |
| C5 | 122 | 121 | 121 | 122 | [124.5]/123.9 |
| C6 | 135 | 135 | 135 | 138 | 140.1 |
| C4′ | 127 | 127 | 128 | 128 | 129.7 |
| C5′ | 123 | 122 | 124 | 122 | [123.9]/124.5 |
| C6′ | 138 | 141 | 139 | 141 | 141.3 |
NMR measurements in solution
The trends predicted by the calculations are confirmed by concentration-dependent 1H NMR measurements on 1c in CDCl3. As shown in Fig. 6 (and also in ESI†, Table S2) the signals of all protons in the rigid part of the molecules shift upfield upon increasing concentration and hence aggregation. The largest shifts occur for the protons at the central benzene, the inner amide and the outer H-6′. This can be explained by a preferred conformation in the aggregated state in which the inner carbonyl groups are out of the plane of the central benzene ring. The smaller shift of protons H-4 and H-4′ may indicate that in comparison, torsions around the bipyridine moiety are less important, which is in agreement with the very strong intramolecular hydrogen bonding. Similar experiments performed at variable temperature show the same trend: when temperature decreases and thereby aggregation increases, there is a generalised upfield shift for all the aromatic signals (see ESI†, Fig. S4).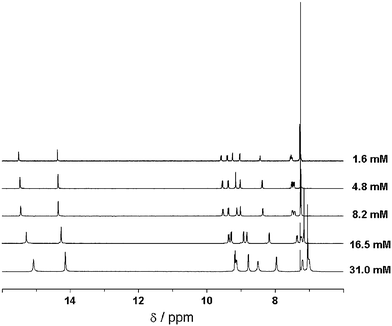
It is important to notice that in both series of experiments, concentration- and temperature-dependent, shifts occur gradually. This behaviour points to an isodesmic mechanism for the self-assembly process.30 The gradual shift of the signals in a broad concentration/temperature range could be explained provided we had in these conditions mixtures of oligomers, whose average length would increase with increasing concentration or decreasing temperature. In contrast, in the cooperative self-assembly mechanism we would expect the shifts taking place in a narrower range, since only long chains and monomers are present when this mechanism rules.30 The NMR results are confirmed by circular dichroism spectroscopy as discussed below. The selection of chloroform-d, a solvent that counteracts aggregation, was justified since less polar solvents such as cyclohexane-d12 only gave rise to very broad absorptions even at elevated temperatures. Moreover, broadening of the peaks and shielding of protons are already observed when raising the concentration in CDCl3 to values above 5 mM.
NMR measurements in the solid state
The upper part of Fig. 5 shows the experimental 1H solid-state NMR spectrum of 1b. Comparing the 1H solid-state NMR chemical shifts with those obtained from solution NMR (see ref. 8 and Table 2) reveals large upfield shifts of more than 1 ppm. Those might be explained either by π–π stacking effects or by different hydrogen bonds in the solid and solution phase. A similar effect of about 1 ppm is also observed for the O–C–H protons not listed in Table 2; the corresponding value in the solution is 4.05 ppm, while the value derived from the solid-state spectrum amounts to 3.2 ppm. The signals of the aromatic protons overlap in the spectrum and could not be easily assigned. However, even for those peaks the packing effects are evident, as in the solid-state spectrum all aromatic resonances occur in the range of 5 to 9 ppm compared to 7.5 to 9.6 ppm in the solution spectrum. To investigate proton–proton proximities a two-dimensional 1H–1H double-quantum (DQ) spectrum was recorded using one rotor period excitation/reconversion time and WATERGATE suppression of the alkyl peak (see Fig. 7).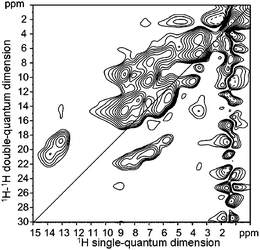 | ||
| Fig. 7 Two dimensional 1H–1H double quantum (DQ)-spectrum of 1b. | ||
In the aromatic region, strong cross peaks originating from neighbouring aromatic protons are observed. Nevertheless, the resolution is still too low for an unambiguous assignment of all protons. Thus, to separate and identify the individual 1H chemical shifts a 1H–13C correlation spectrum was recorded using one rotor period excitation and reconversion time. In such a spectrum, peaks are only observed if the dipole–dipole interaction of the involved nuclei is strong enough, i.e., the distance of the considered nuclei in the range of typical CH bond distances with the further assumption that the molecule is immobile. However, full assignment of all resonances was still not possible, as the 1H resonances show large shifts due to packing effects and the size of these effects for the different protons sensitively depends on the geometry and is, therefore, a priori unknown.
A complete assignment of all 1H and 13C resonances was only possible by combining the calculated chemical shifts with the information derived from the heteronuclear correlation spectrum (see Table 1). This assignment was then verified by reconsidering the 1H–1H DQ spectrum. All cross peaks expected due to close-neighbour contacts in the molecular structure could be identified in the DQ spectrum (e.g., the neighbouring aromatic protons: 4–5, 5–6, 4′–5′, 5′–6′) and all peaks found in this spectrum could be explained by spatial proximities in the structure. The auto peak observed at 6.7 ppm confirms our assignment for the proton of the central aromatic ring, since for two neighbouring molecules the distance of these aromatic protons is only 3.5 Å, whereas the helical arrangement of the molecules in the stack leads for all other protons to larger intermolecular distances. This effect (i.e., larger intermolecular distances due to the helical ordering) is naturally larger for the outer positions. When taking a closer look at the DQ spectrum, two additional weak auto peaks thus are only observed for the inner H-4, outer H-6′ as well as for the inner NH protons, all situated rather close to the center. In a DQ spectrum recorded with a longer excitation time of two rotor periods (not shown here) the intensities of these auto peaks increases, thus again confirming our structure model for 1a and 1b.
While the use of quantum-chemical calculations allows in the present case a complete assignment of both 1H and 13C NMR spectra, it should be noted that, as is often the case, most of the 1H chemical shifts can be assigned without calculations. This, for example, is the case for the inner and outer NH as well as for the ortho H protons. Nevertheless, the ability to assign the complete spectrum convincingly demonstrates the usefulness of quantum-chemical NMR chemical-shift calculations. Please note in addition that the rotation angle φ between discs cannot be predicted by NMR measurements: As was shown by the calculations (vide supra), NMR chemical shifts of the investigated compound do not strongly depend on this parameter. The combination of NMR and quantum-chemical techniques can and does describe the structure of the molecular core, however, and therefore complements the X-ray measurements.
Circular dichroism studies
To confirm the isodesmic mechanism for the self-assembly process, we have used variable-temperature circular dichroism spectroscopy at different concentrations of 1a. In Fig. 8a we show the CD spectra of 1a in methylcyclohexane (MCH) at a concentration of 1.0 × 10−5 M while cooling the solution from 90 to 0 °C. At 90 °C no CD effect is observed as the molecules are molecularly dissolved at this temperature. However, upon decreasing the temperature the solution features a Cotton effect as a result of aggregation and transfer of chirality from the periphery of the individual molecule to the supramolecular architecture.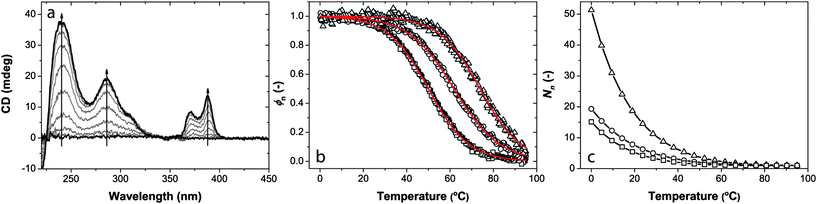 | ||
| Fig. 8 (a) Circular dichroism spectra of 1a recorded in methylcyclohexane (MCH) (c = 1.0 × 10−5 M) at 10 °C intervals while cooling from 90 to 0 °C (arrows indicate decreasing temperature). (b) Degree of aggregation, ϕn, calculated from the molar circular dichroism at 286 nm, as a function of temperature for different concentrations of 1a in MCH: (□) 1.0 × 10−5 M, (○) 5.0 × 10−5 M and (△) 3.5 × 10−4 M, with corresponding fit based on a sigmoidal function. (c) Dependence of stack length, Nn, on temperature at the same concentrations: (□) 1.0 × 10−5 M, (○) 5.0 × 10−5 M and (△) 3.5 × 10−4 M. | ||
For the variable-temperature measurements, we have monitored the molar circular dichroism at 286 nm upon slowly cooling the solution from 95 to 0 °C, for three different concentrations as shown in Fig. 8b. As can be seen in the figure, all curves exhibit a sigmoidal shape as expected for an isodesmic growth mechanism, which confirms the isodesmic behaviour we proposed after our concentration-dependent 1H NMR measurements. Also visible is that with increasing concentration aggregation starts at a higher temperature. For the highest concentration (3.5 × 10−4 M) the molecularly dissolved state is not yet reached at 90 °C. By normalisation of the temperature-dependent molar circular dichroism, the degree of aggregation as a function of temperature was obtained, which we could nicely fit with a sigmoidal function. As the average stack length is directly related to the degree of aggregation31 we were able to determine the average stack length of the stacks at each temperature for the three concentrations. Fig. 8c clearly shows the gradual increase in stack length with decreasing temperature, as is typical for the isodesmic self-assembly mechanism.
Conclusions
The wish to get a detailed molecular picture of supramolecular polymers and systems can only be fulfilled when a large variety of experimental and theoretical approaches is combined. Only together these approaches may provide a convergent answer as is demonstrated for the well-known C3-symmetrical N,N′,N′′-tris[trialkoxybenzoylamino-bipyridinyl] benzenetricarboxamides 1a and 1b.From the revisited X-ray data on 1a and 1b a rotation between superimposed discs in the LC phase has been deduced in the range 13–16° at temperatures above 100 °C but the data cannot unequivocally tell us what would be the tilt angle of the central amides with respect to the central benzene ring. In essence the combination of NMR experiments and quantum-chemical calculations provides a consistent picture of the bowl-shaped molecular core geometry and interactions responsible for stacking. Clearly, no evidence for intermolecular hydrogen-bonding was found and, in fact, for the calculated structure their existence seems highly improbable, as the intermolecular NH⋯O distances are too large. Indeed, as the inter-disc distance amounts only to 3.4 Å, formation of intermolecular H-bonds between N–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O would require a stronger rotation between superimposed units and a stronger tilt of the central amides with respect to the central benzene unit to reconciliate the ∼5.4 Å distance between two hydrogen bonding amides with the inter-disc distance. From the presented calculations it can be concluded that the helical arrangement of the stacks arises from the dynamics in the conformation of the molecules and, hence, from the geometry of the C3-symmetrical core and does not require the presence of additional intermolecular hydrogen-bonds for stabilisation. The dipolar helical alignment of the amide groups may contribute to the stability of the stacks.32
O would require a stronger rotation between superimposed units and a stronger tilt of the central amides with respect to the central benzene unit to reconciliate the ∼5.4 Å distance between two hydrogen bonding amides with the inter-disc distance. From the presented calculations it can be concluded that the helical arrangement of the stacks arises from the dynamics in the conformation of the molecules and, hence, from the geometry of the C3-symmetrical core and does not require the presence of additional intermolecular hydrogen-bonds for stabilisation. The dipolar helical alignment of the amide groups may contribute to the stability of the stacks.32
Concerning the tilt of the central amide functions with respect to the central benzene ring we should admit, that in contrast to our original ideas, the individual molecules are on average more coplanar than those in the stacks: in the latter, propeller formation with concomitant tilt may rigidify the structure. That benzenetricarboxamides can easily deviate from coplanarity by external stimuli is nicely illustrated by the crystal structures of N,N′,N′′-tris(3-pyridyl)33 and N,N′,N′′-tris(2-methoxyethyl)14 benzene-1,3,5-tricarboxamides. This rationalises that stacks are displaying a 15–20 times more intense fluorescence than individual molecules.34 Applying the notions dynamic and rigid for the same system is not paradoxical since the latter refers to femtosecond measurements like fluorescence and the former to actions on > millisecond time scale.
Gaining the correct molecular picture to describe all events during the formation of helical rigid rod aggregates in dilute solution and the subsequent formation of columnar liquid crystals in the bulk by additional lateral interactions for molecules 1a and 1b, has been a challenging task and even now we have to realise that the answer is not unique but one with some degree of freedom. In Fig. 9 a molecular modeling picture of the stacks including the peripheral benzene moieties is presented.
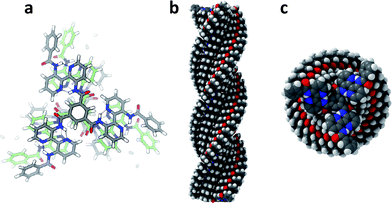 | ||
| Fig. 9 Modeling of stacks of N,N′,N′′-tris(3′-benzoylamino-2,2′-bipyridinyl)benzene-1,3,5-tricarboxamide lacking only the 9-alkoxy tails of 1a and 1b. Parameters: rotation 13°; tilt −20°; bisacylaminobipyridine moieties coplanar; P-helix with carbonyl oxygens down. (a) Ball-and-stick model trimer; (b) filled-space model side view of 28-mer; (c) filled-space model top view of 28-mer. | ||
Adopting a rotation of superimposed molecules by 13° and a tilt of the central amides of −20°, all atoms can be nicely accommodated and, importantly, the peripheral gallic moieties might contribute to the stability of the stacks since superimposed benzene rings are still at stacking distance (an angle of 13° corresponds to a translation of the benzene rings of ∼2.75 Å) especially by their additional out of plane rotation by −30°. This is essential to rationalise the efficiency of chirality transfer from the periphery to the core. Moreover, from this picture a distance of ∼3.65 ± 0.1 Å between the centroids of superimposed bipyridines can be deduced, being very close to the strong additional 3.7 Å reflection observed besides the inter-disc distance of 3.4 Å in the X-ray measurements.
What the study of these supramolecular systems has clearly shown is that inadvertent belief in analogy may offer the wrong conclusions. The analogy with small N,N′,N′′-trialkylbenzene-1,3,5-tricarboxamides was so straightforward that we – without hesitation – were willing to adopt a comparable type of aggregation, especially in view of the analogous response in terms of chirality transfer and expression of “Sergeant-and-soldiers-principle” and “Majority-rules”. Obviously, we did not expect the strong reduction in rotation angle of superimposed units and the transition from a cooperative nucleation-growth to an isodesmic stacking mechanism upon going from small to large benzenetricarboxamides. A lesson that may be drawn from this endeavor is that secondary interactions are subtle and that seemingly small variations in structure may dramatically alter the outcome for a supramolecular structure.
Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft through SFB 625, the Fonds der Chemischen Industrie, and the Council for Chemical Research of the Netherlands Organisation for Scientific Research (CW-NWO) is gratefully acknowledged. The authors are indebted to the personnel of BM26/DUBBLE and especially to Dr G. Portale for assistance during the WAXD experiments at ESRF. Furthermore, NWO (Nederlandse Organisatie voor Wetenschappelijk Onderzoek) and ESRF are acknowledged for granting the beamtime. Professor W. de Jeu is acknowledged for providing us access to the X-ray diffraction equipment at the FOM institute (NL), and Dr D. Byelov for his assistance with the equipment. R. M.-R. acknowledges support from EU through a Marie Curie IntraEuropean Fellowship (Project MEIF-CT-2006-042044) within the EC Sixth Framework Programme. Dr M. A. J. Veld is kindly acknowledged for his help with the artwork.Notes and references
- K. Maeda and E. Yashima, Top. Curr. Chem., 2006, 265, 47–88 CAS.
- D. Pijper and B. L. Feringa, Soft Matter, 2008, 4, 1349–1372 RSC.
- D. Demus, J. Goodby, G. W. Gray, H.-W. Spiess and V. Vill, Handbook of Liquid Crystals, Wiley-VCH, Weinheim, 2002 Search PubMed.
- T. Plesnivy, H. Ringsdorf, P. Schuhmacher, U. Nütz and S. Diele, Liq. Cryst., 1995, 18, 185–190.
- B. Mohr, G. Wegner and K. Ohta, J. Chem. Soc., Chem. Commun., 1995, 995–996 RSC.
- A. Stabel, P. Herwig, K. Müllen and J. P. Rabe, Angew. Chem., Int. Ed. Engl., 1995, 34, 1609–1611 CrossRef CAS.
- H. Kretzschmann, K. Müller, H. Kolshorn, D. Schollmeyer and H. Meier, Chem. Ber., 1994, 127, 1735–1745 CAS.
- J. Zhang and J. S. Moore, J. Am. Chem. Soc., 1994, 116, 2655–2656 CrossRef CAS.
- S. Bauer, T. Plesnivy, H. Ringsdorf and P. Schuhmacher, Makromol. Chem., Macromol. Symp., 1992, 64, 19–32 Search PubMed.
- C. Mertesdorf and H. Ringsdorf, Mol. Eng., 1992, 2, 189–212 CrossRef CAS.
- A. R. A. Palmans, J. A. J. M. Vekemans, H. Fischer, R. A. Hikmet and E. W. Meijer, Chem.–Eur. J., 1997, 3, 300–307 CrossRef CAS.
- A. R. A. Palmans, J. A. J. M. Vekemans, E. E. Havinga and E. W. Meijer, Angew. Chem., Int. Ed. Engl., 1997, 36, 2648–2651 CrossRef CAS.
- J. van Gestel, A. R. A. Palmans, B. Titulaer, J. A. J. M. Vekemans and E. W. Meijer, J. Am. Chem. Soc., 2005, 127, 5490–5494 CrossRef CAS.
- M. P. Lightfoot, F. S. Mair, R. G. Pritchard and J. E. Warren, Chem. Commun., 1999, 1945–1946 RSC.
- P. P. Bose, M. G. B. Drew, A. K. Das and A. Banerjee, Chem. Commun., 2006, 3196–3198 RSC.
- L. Brunsveld, A. P. H. J. Schenning, M. A. C. Broeren, H. M. Janssen, J. A. J. M. Vekemans and E. W. Meijer, Chem. Lett., 2000, 292–293 CrossRef CAS.
- A. J. Wilson, J. van Gestel, R. P. Sijbesma and E. W. Meijer, Chem. Commun., 2006, 4404–4406 RSC.
- M. M. J. Smulders, P. J. M. Stals, T. Mes, T. F. E. Paffen, A. P. H. J. Schenning, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2010, 132, 620–626 CrossRef CAS.
- M. M. J. Smulders, I. A. W. Filot, J. M. A. Leenders, P. van der Schoot, A. R. A. Palmans, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2010, 132, 611–619 CrossRef CAS.
- D. Durand, J. Doucet and F. Livolant, J. Phys. II, 1992, 2, 1769–1783 CrossRef CAS.
- K. Nagai, K. Sakajiri, K. Maeda, K. Okoshi, T. Sato and E. Yashima, Macromolecules, 2006, 39, 5371–5380 CrossRef CAS.
- J. Barberá, E. Cavero, M. Lehmann, J.-L. Serrano, T. Sierra and J. T. Vazquez, J. Am. Chem. Soc., 2003, 125, 4527–4533 CrossRef CAS.
- E. Fontes, P. A. Heiney and W. H. de Jeu, Phys. Rev. Lett., 1988, 61, 1202 CrossRef CAS.
- X. Feng, W. Pisula, M. Takase, X. Dou, V. Enkelmann, M. Wagner, N. Ding and K. Müllen, Chem. Mater., 2008, 20, 2872–2874 CrossRef CAS.
- J. Barberá, J. Jiménez, A. Laguna, L. Oriol, S. Pérez and J. L. Serrano, Chem. Mater., 2006, 18, 5437–5445 CrossRef CAS.
- M. Lehmann, M. Jahr, F. C. Grozema, R. D. Abellon, L. D. A. Siebbeles and M. Müller, Adv. Mater., 2008, 20, 4414–4418 CrossRef CAS.
- W. Cochran, F. H. Crick and V. Vand, Acta Crystallogr., 1952, 5, 581–586 CrossRef CAS.
- W. Pisula, Ž. Tomović, M. D. Watson, K. Müllen, J. Kussmann, C. Ochsenfeld, T. Metzroth and J. Gauss, J. Phys. Chem. B, 2007, 111, 7481–7487 CrossRef CAS.
- C. Ochsenfeld, S. P. Brown, I. Schnell, J. Gauss and H. W. Spiess, J. Am. Chem. Soc., 2001, 123, 2597–2606 CrossRef CAS.
- T. F. A. de Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS.
- M. M. J. Smulders, M. M. L. Nieuwenhuizen, T. F. A. de Greef, P. van der Schoot, A. P. H. J. Schenning and E. W. Meijer, Chem.–Eur. J., 2010, 16, 362–367 CrossRef CAS.
- A. Choudhary, D. Gandla, G. R. Krow and R. T. Raines, J. Am. Chem. Soc., 2009, 131, 7244–7246 CrossRef CAS.
- A. R. A. Palmans, J. A. J. M. Vekemans, H. Kooijman, A. L. Spek and E. W. Meijer, Chem. Commun., 1997, 2247–2248 RSC.
- L. Brunsveld, H. Zhang, M. Glasbeek, J. A. J. M. Vekemans and E. W. Meijer, J. Am. Chem. Soc., 2000, 122, 6175–6182 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, additional X-ray and NMR data. See DOI: 10.1039/c0sc00292e |
| This journal is © The Royal Society of Chemistry 2011 |