Triplet–triplet annihilation based upconversion: from triplet sensitizers and triplet acceptors to upconversion quantum yields
Jianzhang
Zhao
*,
Shaomin
Ji
and
Huimin
Guo
State Key Laboratory of Fine Chemicals, School of Chemical Engineering, Dalian University of Technology, Dalian, 116024, P. R. China. E-mail: zhaojzh@dlut.edu.cn; Fax: + 86 (0)411 8498 6236; Web: http://finechem.dlut.edu.cn/zhaojianzhang/guohm/index.htm
First published on 5th October 2011
Abstract
Triplet–triplet annihilation (TTA) is a promising upconversion approach due to its low excitation power density (solar light is sufficient), high upconversion quantum yield, readily tunable excitation/emission wavelength and strong absorption of excitation light. This review focuses on the reported TTA based upconversion examples, the challenges that are facing the developments of TTA upconversion and the design rationales for the triplet sensitizers and triplet acceptors.
 Jianzhang Zhao | Prof. Jianzhang Zhao received his Ph.D. degree at Jilin University in 2000. Then he carried out postdoctoral research at POSTECH (Pohang, South Korea), Max-Planck Research Unit (Halle, Germany) and University of Bath (Bath, U.K.) during 2000–2005. He took up his current position in 2005. His research interests involve fluorescent molecular probes and phosphorescent transition metal complexes, ranging from synthesis to study of photophysical properties with steady-state and time-resolved spectroscopy, as well as application of DFT calculations in luminescence studies. |
 Shaomin Ji | Shaomin Ji received her B. S. degree in Applied Chemistry from Hebei University of Technology in 2006. Since 2006 she has been a Ph.D. student in Prof. J. Zhao's group, at Dalian University of Technology. Currently she is working on fluorescent molecular probes and phosphorescent Ru(II) complexes, including synthesis and study of the photophysical properties with steady-state and time-resolved spectroscopy and application of the complexes for luminescent O2 sensing and triplet–triplet annihilation upconversions. |
 Huimin Guo | Dr Huimin Guo received her Ph.D. degree in theoretical and physical chemistry under the direction of Prof. Bernhard Dick in 2005 from the University of Regensburg, Germany. She then became a lecturer in the School of Chemistry, Dalian University of Technology, and was promoted to associate professor in 2010. Her research interests mainly focus on fluorescent molecular probes, and photochemical and photophysics of phosphorescent transition metal complexes. |
1. Introduction
Upconversion, that is, observation of photon emission, or more generally, population of excited state at higher energy (shorter wavelength) with excitation at lower energy (longer wavelength), has attracted much attention due to its potential applications for photovoltaics, artificial photosynthesis, photocatalysis, and optics, etc.1–4 For example, it is difficult for the dye-sensitized solar cell (DSCs) to utilize the solar light in the near-IR region, despite the power of the solar light in this wavelength range being intense. The efficiency of the DSCs can be improved with upconversion materials that can convert the radiation at a longer wavelength into radiation at a shorter wavelength.Currently, a few techniques are available for upconversion, including upconversion with two-photon absorption dyes (TPA), upconversion with inorganic crystals (such as KDP), and rare earth metarials,1–4etc. However, these techniques usually suffer from drawbacks of high excitation power, poor absorption of visible light, and low upconversion quantum yield, etc. Therefore, these techniques are unlikely to be used for applications with a light source at low excitation power density, such as solar light. For example, a coherent laser with high power-density (typically MW cm−2) is required for excitation of TPA dyes, which is well beyond the energy of a normal light source (the power-density of the terrestrial solar irradiance is ca. 0.10 W cm−2, AM1.5G). Furthermore, from a chemist's perspective, it is difficult to tailor the structure of TPA dyes to achieve a specific upconversion wavelength and at the same time, to maintain a high TPA cross section. Recently, the upconversion schemes with rare earth materials have attracted much attention. However, the absorption of these materials are usually weak, thus the overall upconversion capability (η = ε × ΦUC, ε is the molar extinction coefficient of the upconversion materials at the excitation wavelength and ΦUC is the upconversion quantum yield) is poor.
Recently, a new upconversion scheme based on triplet–triplet annihilation has been developed and has attracted much attention.5–10 TTA upconversion shows advantages over the aforementioned upconversion techniques. For example, the excitation power density required for TTA upconversion is quite low and the excitation need not be coherent. Excitation with energy density of a few mW cm−2 is sufficient to sensitize the upconversion process.7 Thus it is possible to use solar light as the excitation source for TTA upconversion. Furthermore, the excitation wavelength and emission wavelength of TTA upconversion can be readily changed, simply by independent selection of the two components of the upconversion scheme, i.e. the triplet sensitizer and the triplet acceptor (annihilator/emitter) (but the energy levels of the excited state of the sensitizers and the acceptors must be matched. See Scheme 1 and later section for detail). Thus the TTA upconversion is promising for applications such as photovoltaics, photocatalysis, and many other light-driven photophysical and photochemical processes. Recently a review on the subject of TTA upconversion was reported, but it was much focused on the work from Castellano's research group.8 Herein we make an attempt to cover a wider range of the related research.
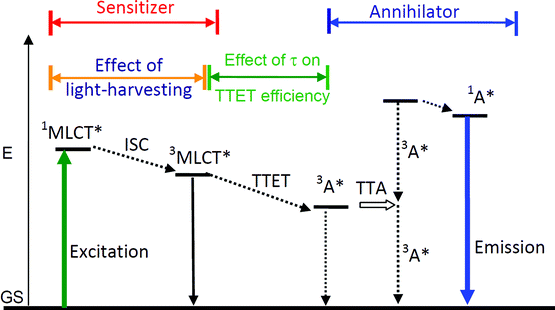 | ||
| Scheme 1 Qualitative Jablonski Diagram illustrating the sensitized TTA upconversion process between triplet sensitizer and acceptor (annihilator/emitter). The effect of the light-harvesting ability and the excited state lifetime of the sensitizer on the efficiency of the TTA upconversion is also shown. E is energy. GS is ground state (S0). 3MLCT* is the metal-to-ligand-charge-transfer triplet excited state. TTET is triplet–triplet energy transfer. 3A* is the triplet excited state of annihilator. TTA is triplet–triplet annihilation. 1A* is the singlet excited state of annihilator. The emission band observed for the sensitizers alone is the 3MLCT emissive excited state. The emission bands observed in the TTA experiment are the simultaneous 3MLCT* emission (phosphorescence) and the 1A* emission (fluorescence). | ||
Different from most of the other upconversion methods, the TTA upconversion is based on mixing the triplet sensitizer and triplet acceptor (annihilator/emitter) together. The excitation energy is harvested by the sensitizer and the energy is transferred to the acceptor via triplet–triplet energy transfer (TTET), which will give emission at higher energy level than the excitation. The photophysics of TTA upconversion can be illustrated by a Jablonski diagram (Scheme 1).
Firstly the sensitizer is excited with photo-irradiation (Scheme 1). The singlet excited state will be populated (S0→S1). Then with intersystem crossing (ISC, for example S1→T1), in which the heavy atom effect of transition metal atom is often required, the triplet excited state of the sensitizer will be populated. It should be noted that direct excitation into the T1 state is forbidden (S0→T1 is usually a forbidden process). Since the lifetime of the triplet excited state is much longer than that of the singlet excited state, thus the energy can be transferred from the triplet sensitizer to the triplet acceptor, via the TTET process. Note the energy transfer between the triplet states is usually a Dexter process and it requires contact of the two components.11 The triplet acceptor molecules at the triplet excited state will collide with each other and produce the singlet excited state of the acceptor, follow the spin statistic law eqn (1). The radiative decay from the singlet excited state of acceptor produces the upconverted fluorescence, for which the energy is higher than the excitation light. One example is illustrated in Fig. 1.
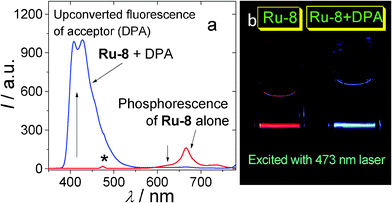 | ||
| Fig. 1 Illustration of the TTA upconversion. (a) Emission of the complex Ru-8 (see Fig. 4 for molecular structure) and the upconversion with Ru-8 as sensitizer and DPA as acceptor (see Fig. 2 for molecular structure). Excited by 473 nm laser. The asterisk indicates laser scattering. (b) Photographs of the upconversion samples of (a). | ||
Based on the spin-statistic law eqn (1), the limit for the efficiency of the TTA upconversion is 11.1%.8 However, examples that exceed this limit have been reported, indicating that excited states other than the triplet states can also lead to the singlet excited states.8
The spin manifold of the triplet encounters of the acceptors was governed by the so-called spin-statistical factors.12,13 When two excited triplets (3A1) interact, nine encounter-pair spin states are produced with equal probability which is composed of three distinct sublevels, five of which are quintet, three are triplet, and one is singlet. Thus, spin statistics predicts that singlet productor represents 1/9 (or 11.1%) of the annihilation events. Thus the maximal upconversion quantum yield will be no higher than 11.1% eqn (2), given that both the quenching efficiency Φq and the fluorescence quantum yield of acceptor ΦF are 100%.12 While most of the TTA upconversion show a quantum yield less than 10%,3,4 however, a few recently reported cases (including the results from our laboratory) do show that upconversion quantum yield higher than 11.1% were observed.9 These results suggest that the triplets and possibly the quintets are also leading to upconversion.
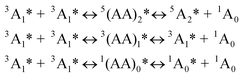 | (eqn 1) |
| ΦUC = Φq × ΦTTA × ΦF | (eqn 2) |
For example, it was proposed that the quintet state 5A* will have a 92% chance to decay into two molecules at the triplet excited state (3A*),12 which are then involved in the TTA again. Thus, the maximal upconversion quantum yield is definitely higher than the previously thought 11.1%.
The upconversion quantum yield can be described by eqn (2), where Φq is the energy transfer efficiency (TTET), ΦTTA is the efficiency to produce singlet excited state by the TTA process, and ΦF is the fluorescence quantum yield of the acceptor.
It should be pointed out that several photophysical parameters of the sensitizer and acceptor are crucial for TTA upconversion. (1) The light harvesting ability of the triplet sensitizer. Usually transition metal complexes show weak absorption in the visible region and this is detrimental to the applications of TTA upconversion. TTA upconversion requires that the concentration of the sensitizers at the triplet excited state be high, thus the acceptor molecules at the triplet excited state will be high, and the TTA upconversion will be more significant.7 The reason for this mandate is the bimolecular feature of the TTET and the TTA processes. With higher concentration of the triplet sensitizers at the triplet excited state, the TTET process will be more efficient to produce the acceptors at triplet excited state. Herein we propose that the overall upconversion capability (η) of a triplet sensitizer can be better evaluated by ε × ΦUC, i.e. not only the ΦUC value eqn (5). (2) The triplet excited state quantum yield of the sensitizer must be high because it is the triplet excited state, not the singlet excited state that directly produced upon photoexcitation, that is involved in the critical TTET process. Most triplet sensitizers are transition metal complexes, for which the ISC process is very often with unit efficiency (ΦISC is close to 100%). (3) The lifetime of the triplet excited state of the sensitizers should be long. Long-lived T1 excited state of the sensitizer will lead to a more efficient TTET process because the TTET process is actually a two-molecular quenching procedure, long-lived T1 excited state of the sensitizer will increase the diffusion distance and make the encounter of the sensitizer and the acceptor more likely. However, although some of the triplet sensitizers show long-lived T1 excited state, but usually the T1 excited state of the transition metal complexes are short (in a few microseconds range). (4) The relative energy levels of the triplet sensitizers and the triplet acceptors must be appropriate to maximize the TTET efficiency. (5) The T1 excited state energy level and the S1 excited state energy level of the triplet acceptor fulfil the relation 2 × ET1 > ES1, where ET1 is the energy level of the T1 excited state and the ES1 is the energy level of the S1 excited state. (6) The radiative decay of the S1 excited state should be efficient to produce intense upconverted fluorescence emission, i.e. the fluorescence quantum yield of the acceptor (ΦF, eqn (2)) should be high.
Following these photophysical mandates, the design rationales of the triplet sensitizers and the acceptors can be summarized as, (1) triplet sensitizer should be with strong absorption at the excitation wavelength (large ε values); (2) efficient ISC to produce the T1 excited state; (3) the lifetime of the T1 excited state of the sensitizer must be long; (4) the energy levels of the excited states of the sensitizers and the acceptors must be matched in order to enhance the TTET process.
The TTA upconversion can be quantitatively described with two parameters, i.e. the efficiency of TTET process and the upconversion quantum yields (ΦUC). The TTET efficiency can be measured by the quenching experiments, with the triplet acceptor as the quencher. Fitting the quenching result with the Stern–Volmer equation will give the KSV value and the bimolecular quenching constants kqeqn (3),
| I0/I = 1 + KSV[Q], KSV = kq × τ0 | (eqn 3) |
where τ0 is the lifetime of the triplet excited state of the sensitizer and [Q] is the concentration of the quenchers at which the I (residual emission of the sensitizer) is determined. It should be noted that in some cases the triplet sensitizer is non-phosphorescent, thus the quenching can be measured by the variation of the lifetime of the T1 excited state of the sensitizer, such as by using time-resolved transient absorption spectroscopy.
Experimentally the upconversion quantum yield (ΦUC) can be determined by eqn (4),8 where Φunk, Aunk, Iunk and ηunk represent
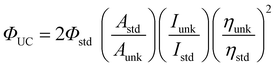 | (eqn 4) |
the quantum yield, absorbance, integrated photoluminescence intensity of the samples and the refractive index of the solvents, respectively. Since two photons are required to generate one upconverted photon, in order to keep the maximum quantum yield as a unit, the equation is multiplied by a factor of 2.8
| η = ε × ΦUC | (eqn 5) |
Herein we propose to use the ε × ΦUC to evaluate the overall upconversion capability (η) of a triplet sensitizer eqn (5), where ε is the molar extinction coefficient of the upconversion materials and ΦUC is the upconversion quantum yield determined with eqn (4). Triplet sensitizers with large η values are more likely to be ideal for practical applications. On the contrary, materials with large ΦUC value but small ε value are not ideal for applications. Furthermore, it should be pointed out that TTA upconversion schemes with excitation at the red or near-IR region are ideal for applications, such as photovoltaics.
We noted that much room is left for the development of the TTA upconversion. For example, currently the triplet sensitizers are limited to the Pt(II)/Pd(II) porphyrin complexes and the upconversion quantum yield is only moderate. The molecular design rationales disclosed above will be helpful for the development of the TTA upconversion.
2. Triplet sensitizers for TTA upconversion
2.1 Ru(II) polymine complexes as triplet sensitizers
Ru(II) polyimine complexes have been investigated for a long time and these complexes have been extensively used in photovoltaics, molecular arrays, etc. The most significant photophysical features of the Ru(II) polyimine complexes are population of the triplet excited states upon photoexcitation, moderate absorption in the visible range, long-lived T1 excited state, and efficient ISC (the quantum yield of the S1→T1 is close to unity). These features are ideal for application of the complexes as triplet sensitizers for TTA upconversion.In 2005, Castellano et al. used [Ru(dmb)3][PF6]2 (Ru-1) to sensitize the TTA upconversion, with 9,10-diphenylanthracene (DPA) as triplet acceptor (Fig. 2).14 The absorption of the complex is peaked at ca. 450 nm. The energy level of the triplet excited state of the sensitizer can be easily derived from the phosphorescence wavelength, at ca. 600 nm (2.07 eV). Thus, DPA (A-1 in Fig. 2) was used as the triplet acceptor, for which the energy level of the T1 excited is 1.77 eV (700 nm). The mixed solution of [Ru(dmb)3]2+ and DPA was excited with a green laser (λex = 514.5 nm, 24 mW or λex = 532 nm, <5 mW), the upconverted blue fluorescence emission of DPA was observed (the upconverted fluorescence is centered at 430 nm, thus the anti-Stokes shift is ca. 100 nm). This result demonstrated that the TTA upconversion can be achieved with low-power density irradiation. However, no efficiency of the TTET process and the quantum yield of the upconversion were reported for Ru(dmb)3[PF6]2.14
![Triplet sensitizer [Ru(dmb)3]2+ (Ru-1, dmb = 4,4′-dimethyl-2,2′-bipyridine) and the triplet sensitizer 9,10-diphenylanthracene (DPA, A-1) for triplet–triplet annihilation based upconversion.](/image/article/2011/RA/c1ra00469g/c1ra00469g-f2.gif) | ||
| Fig. 2 Triplet sensitizer [Ru(dmb)3]2+ (Ru-1, dmb = 4,4′-dimethyl-2,2′-bipyridine) and the triplet sensitizer 9,10-diphenylanthracene (DPA, A-1) for triplet–triplet annihilation based upconversion. | ||
An intramolecular approach, i.e.Ru(II) complexes with covalently linked anthracene moiety as the integrated sensitizer/acceptor [Ru(dmb)2(bpy-An)] (dmb is 4,4′-dimethyl-2,2′-bipyridine and bpy-An is 4-methyl-4′-(9-anthrylethyl)-2,2′-bipyridine) was reported.15 The results show that the upconversion is more efficient for the intramolecular approach than the intermolecular method (enhanced by 2.9-fold).15
It should be pointed out that TTA upconversion is dependent on many factors, such as the concentration of triplet sensitizer and acceptor, the power density of the excitation, etc. The success of using Ru(dmb)3[PF6]2 to achieve upconversion with low-power density excitation is attributed to the long lifetime of the 3MLCT excited state of the complex (τ = 0.87 μs). The prolonged T1 excited state lifetime is beneficial to improve the TTET process and, as a result, can lead to more DPA molecules at the singlet excited state.
However, upconversion with Ru(dmb)3[PF6]2 suffers from some drawbacks. Firstly, the UV-vis absorption of the Ru(II) complex is weak in the visible range and the absorption is limited <450 nm, this is typical for normal Ru(II) diimine complexes. Triplet sensitizers with intensive absorption in longer wavelength are desired. Second, the lifetimes of Ru(II) complexes are less than 1 μs. Longer lifetimes will enhance the TTET process, which has been demonstrated in luminescent O2 sensing, for which the key photophysical step is also the TTET process.16–18 Thus, it is desired to develop triplet sensitizers with prolonged T1 excited state lifetimes.
One method to access the long-lived 3MLCT excited state of Ru(II) polyimine complexes is to optimize the coordination geometry of the Ru(II) centre, by selection of appropriate ligands. For example, the Ru-2 ([Ru(tpy)2]2+) shows a T1 excited state lifetime of 0.25 ns (Fig. 3).19,20 With bpy ligand (Ru-3), the T1 lifetime was prolonged to 1.0 μs.21 By optimization of the geometry of the NˆNˆN ligand (Ru-4), the lifetime of the 3MLCT excited state was extended to 3.0 μs.21 However, we propose that a much longer lived T1 excited state is necessary to enhance the TTET, and thus the TTA upconversion significantly.
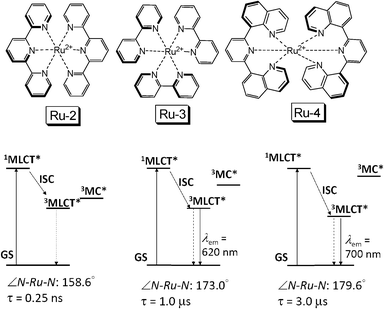 | ||
| Fig. 3 Molecular structures of typical Ru(II) polyimine complexes Ru-2, Ru-3 and Ru-4. The bottom panel shows the simplified energy level diagrams and the emission states for Ru-2, Ru-3 and Ru-4, respectively. | ||
It has been shown that the 3IL (intraligand) excited state of the Ru(II) complexes show a much longer lifetime than the 3MLCT excited state.22–25 Previously we demonstrated that the O2 sensing property of the complexes can be significantly improved with the long-lived 3IL excited state.16–18 Since the critical photophysical process involved in the luminescent O2 sensing (TTET) is similar to that for the TTA upconversion (Scheme 1), thus we envisaged that the Ru(II) polyimine complexes with the long-lived 3IL excited state can be used for TTA upconversion.
Recently we prepared two Ru(II) complexes Ru-7 and Ru-8 that showed long-lived T1 excited states (Fig. 4).16 Complex Ru-8 showed a much longer T1 excited state lifetime (108.0 μs, previously it was determined as 58.4 μs) than the model complex Ru-5 (0.45 μs). Thus we envisaged that the TTET process with Ru-4 as the triplet sensitizer would be much more efficient than that with Ru-5 as the triplet sensitizer. With DPA as the triplet acceptor, we investigated the TTA upconversion with the complexes as triplet sensitizers.26
![Chemical structures of the sensitizer RuII complexes Ru-5 ∼ Ru-8. Note the complexes are dications and the [PF6]− ions are omitted for clarity. The compounds are from ref. 16.](/image/article/2011/RA/c1ra00469g/c1ra00469g-f4.gif) | ||
| Fig. 4 Chemical structures of the sensitizer RuII complexes Ru-5 ∼ Ru-8. Note the complexes are dications and the [PF6]− ions are omitted for clarity. The compounds are from ref. 16. | ||
The absorption and phosphorescence of Ru-5 and Ru-8 were compared (Fig. 5). Ru-8 gives a more intense absorption than the model complex Ru-5. The typical 3MLCT emission band was observed for Ru-5 (structureless). For Ru-8, however, the structured 3IL emission band was observed. Ru-7 also showed a prolonged lifetime compared to the model complex Ru-5, due to the 3MLCT/3IL excited state equilibrium.16
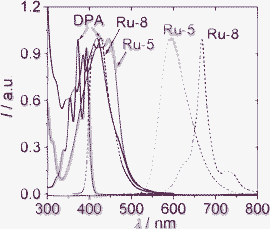 | ||
| Fig. 5 Normalized absorbance (solid lines) and emission spectra (dotted lines) of DPA, Ru-5 and Ru-8 in acetonitrile (1.0 × 10−5 M). DPA, λex = 380 nm; Ru-5, λex = 446 nm; Ru-8, λex = 418 nm. 25 °C. Adapted from ref. 26 with permission. | ||
In the presence of triplet acceptor DPA, the phosphorescence of the sensitizers was quenched to different extents (Fig. 6b). At the same time, the upconverted blue emission of DPA was observed in the region of 400 nm–550 nm. The upconversion is most significant for Ru-7 (ΦUC = 9.8%) and Ru-8 (ΦUC = 9.6%).26 The more efficient TTA upconversion with Ru-7 and Ru-8 than that with the model complexes Ru-5 (ΦUC = 0.9%) and Ru-6 (ΦUC = 4.5%) is attributed to the long-lived T1 excited state of Ru-8 and Ru-7, with which the critical process of the TTA upconversion, that is, the TTET process, was enhanced. Under similar conditions [Ru(dmb)3]2+ gives ΦUC value of 1.0%. The TTET process of the TTA upconversion can be quantitatively evaluated by the quenching of phosphorescence of sensitizers with acceptor DPA (Fig. 7). The largest Stern–Volmer quenching constant was observed for Ru-8 (9.93 × 105 M−1). The KSV value of Ru-8 is much larger than that of Ru-5 (4.59 × 103 M−1) and Ru-6 (1.47 × 104 M−1). A large value was also observed for Ru-7 (τ = 9.22 μs. KSV = 1.70 × 105 M−1).
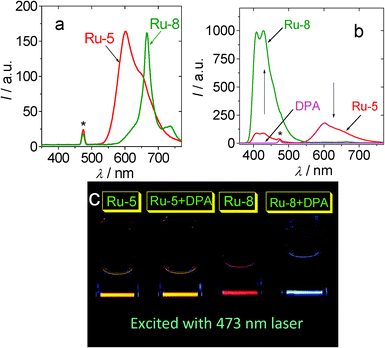 | ||
| Fig. 6 Emission and upconversion of Ru-5 and Ru-8 with 473 nm laser excitation. (a) Emission of the Ru(II) complexes. Excited by blue laser (λex = 473 nm, 5 mW). In order to show the different emission intensity of the complexes, the spectra were not normalized. (b) The upconverted DPA fluorescence and the residual phosphorescence of the mixture of DPA (4.3 × 10−5 M) and Ru-5 or Ru-8, respectively. (c) The photographs of the upconversion (samples of a and b). In deaerated CH3CN solution. The complexes solution are 1.0 × 10−5 M. The asterisks in (a) and (b) indicate the scattered 473 nm excitation laser. 25 °C. Adapted from ref. 26 with permission. | ||
![Stern–Volmer plots generated from intensity quenching of complex [Ru(dmb)3]2+ (λex = 460 nm), Ru-5 (λex = 446 nm), Ru-6 (λex = 450 nm), Ru-7 (λex = 450 nm) and Ru-8 (λex = 418 nm). Phosphorescence measured as a function of DPA concentration in CH3CN. 1.0 × 10−5 mol dm−3. 25 °C. Reproduced with permission from ref. 26.](/image/article/2011/RA/c1ra00469g/c1ra00469g-f7.gif) | ||
| Fig. 7 Stern–Volmer plots generated from intensity quenching of complex [Ru(dmb)3]2+ (λex = 460 nm), Ru-5 (λex = 446 nm), Ru-6 (λex = 450 nm), Ru-7 (λex = 450 nm) and Ru-8 (λex = 418 nm). Phosphorescence measured as a function of DPA concentration in CH3CN. 1.0 × 10−5 mol dm−3. 25 °C. Reproduced with permission from ref. 26. | ||
The upconversion quantum yield with Ru-8 as the sensitizer is much higher than that with Ru-5 or Ru(dmb)3 as the sensitizers. We noticed that the upconversion of Ru-8 is not significantly higher than that of Ru-7, despite the much longer T1 lifetime of Ru-8 than that of Ru-7. We attributed this apparently small upconversion quantum yield to the lower T1 excited state energy level of Ru-8.
It should be pointed out that the absorption of the typical Ru(II) polyimine complexes are weak in the visible region and the T1 excited state lifetime is not long (usually less than 1 μs). Thus, another kind of transition metal complexes, i.e. the Pt(II)/Pd(II) porphyrin complexes have been used for TTA upconversion.
2.2 Pt(II) Pd(II) porphyrin complexes
Pt(II) porphyrin complexes, such as PtOEP (Pt-1, OEP = octaethylporphyin) have been used in luminescent oxygen sensing and photodynamic therapy, both applications are based on the capability of visible light absorption and population of triplet excited state upon photoexcitation. Different from the typical Ru(II) polyimine complexes, the Pt(II) porphyrin complexes show intense absorption of visible light and the lifetimes of the T1 excited state of these complexes are much longer, usually longer than 50 μs.27,28Pt(II) porphyrin complexes are triplet emitters with moderate absorption in the green. For example, the Pt-1 (Fig. 8) shows absorption at ca. 400 nm with ε =1.0–5.0 × 105 M−1 cm−1. But usually the absorption at a longer wavelength, i.e. 530 nm, is much weaker. Triplet state energy of the porphyrin complexes is 1.33–1.93 eV (641–930 nm). Thus, Pt-1 was used with DPA (with T1 energy of 1.77 eV, or 700 nm) for non-coherently excited annihilation upconversion.29 It should be pointed out that the complex Pt-1, or other Pt porphyrin complexes, usually show a much longer triplet excited state lifetime than the Ru(II) polyimine complexes. The long-lived triplet excited state of the sensitizer is beneficial for the TTET process and the TTA upconversion.
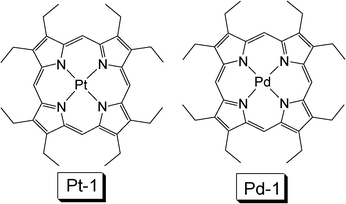 | ||
| Fig. 8 Molecular structures of platinum and palladium octoethylporphyrin complex Pt-1 and Pd-1.11 | ||
In 2006, Baluschev reported TTA upconversion with focused solar light as the excitation source, the external efficiency was 1% (the excitation power density is 10 W cm−2).5Pd-1 (PdOEP) was used as a triplet sensitizer and DPA was used as a triplet acceptor (Fig. 8).
In 2007, Castellano demonstrated that the Pt-1/DPA upconversion scheme is effective even in polymer films with low glass transition temperature. Excitation was at 544 nm.29 It is significant that the upconversion works in the solid matrix in an aerobic atmosphere and with excitation at a low excitation power density of 6–27 mW cm−2. This result paved the way for practical application of the TTA upconversion.
In order to use red light to perform the upconversion, a triplet sensitizer with red light absorption has to be used. Red absorbing sensitizer platinum(II) tetraphenyltetrabenzoporphyrin (Pt-2) (Fig. 9) and palladium porphyrin complex Pd-1 were used as triplet sensitizers (Fig. 8).30–33
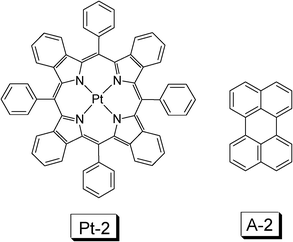 | ||
| Fig. 9 Molecular structures of triplet sensitizer platinum(II) tetraphenyltetrabenzoporphyrin (Pt-2) and the triplet acceptor A-2.30 | ||
The platinum(II) tetraphenyltetrabenzoporphyrin complex Pt-2 (Fig. 9) shows strong absorption at 430 nm (Soret band) and an absorption at 611 nm (Q-band). The complex shows phosphorescence at 770 nm (1.61 eV, τ = 41.5 μs). DPA is not an appropriate triplet acceptor in this case, due to its unmatched triplet state (T1) energy level (1.77 eV), which is higher than the sensitizer and the TTET from the sensitizer to the acceptor will be frustrated. Perylene (A-2) was selected as the triplet acceptor, for which the T1 state energy level is 1.53 eV (Fig. 9). The UV-vis absorption of perylene is in the region <450 nm. With red excitation (635 nm laser), the blue/green fluorescence of perylene was observed. The upconversion quantum yield (ΦUC) is 0.65%.30 Several factors may be responsible for the low upconversion quantum yield, such as the large size of the sensitizer molecule, which may reduce the diffusion ability of the sensitizer at triplet excited state, which is detrimental to the TTET efficiency.11
For applications such as DSCs, the challenge is to effectively harvest the energy of the solar light in the red/near-IR region, where the normal organic dyes show poor absorption. Recently a palladium porphyrin complex that shows absorption in the near-IR region was used for TTA based upconversion (Fig. 10).31 The complex shows intense absorption at ca. 700 nm and the phosphorescence is at 916 nm/942 nm. The near-IR (NIR) absorption of the Pd(II) complex is in particular significance since the low-energy NIR light of the solar irradiance can be utilized with this sensitizer. However, as the energy level of the T1 state of the complex is very low, thus a triplet acceptor with tailored design with matched T1 energy level have to be used (A-3, Fig. 10), which shows green fluorescence emission in the region of 490 nm–600 nm. However, the energy level of the triplet excited state of this compound was not given, but presumably lower than 1.32 eV (942 nm). With excitation at 700 nm, green emission of the acceptor/annihilator in the range of 490 nm–600 nm was observed. The external upconversion quantum yield was determined as 4%.
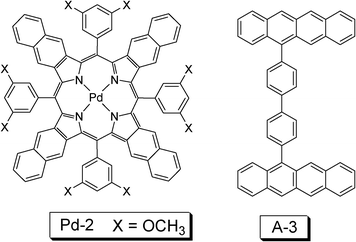 | ||
| Fig. 10 Molecular structures of palladium complex Pd-2 and the triplet acceptor A-3 used for the upconversion. The compounds are from ref. 31. | ||
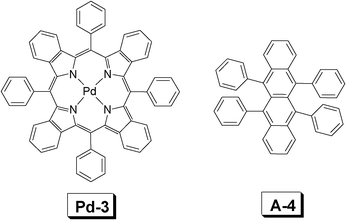 | ||
| Fig. 11 Molecular structures of Pd-3 (sensitizer) and rubrene A-4 (acceptor). The compounds are from ref. 32. | ||
The authors also performed the upconversion with focused solar irradiance. This is particularly interesting because the efficiency of the DSCs may be improved with the NIR absorbing upconversion schemes.
In order to harvest a broad excitation wavelength, two sensitizers were simultaneously used for TTA upconversion.32 The two complexes used as triplet sensitizers are Pd-2 and Pd-3, respectively (Fig. 10, Fig. 11). The two complexes give absorption at 630 nm and 700 nm, respectively. Thus with the sun light as the excitation source, upconversion was observed with rubrene (A-4, Fig. 11) as the triplet acceptor.
In 2010, Castellano reported a TTA upconversion with NIR absorbing triplet sensitizer Ru-9 (Fig. 12).34 The excitation was carried out at 780 nm, and the upconverted emission of the peryleneimide (A-5) is at 541 nm. The upconversion quantum yield was determined as (0.75 ± 0.02)%.
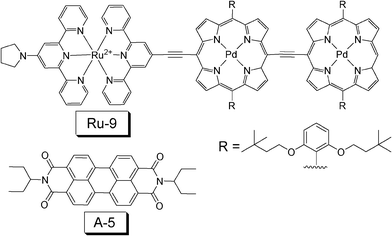 | ||
| Fig. 12 Molecular structures of the near-IR absorbing sensitizer Ru-9 and the triplet acceptor A-5. The compounds are from ref. 34. | ||
Although the Pt(II)/Pd(II) porphyrin complexes have been successfully used as triplet sensitizers for TTA upconversion, we noticed the limitations of these complexes, i.e., the absorption/emission wavelength of the Pt(II)/Pd(II) complexes can not be readily changed by chemical modification of molecular structures of the sensitizers. Thus, it is desired that an alternative type of sensitizer can be developed that shows a tunable excitation/emission wavelength.8 We propose that Pt(II) acetylide complexes will be the choice to address this challenge.
2.3 Pt(II) acetylide complexes as the triplet sensitizers: tunable photophysical properties
Pt(II) acetylide complexes are usually phosphorescent at room temperature and the fluorescence of the ligands are completely quenched in the complexes, indicating efficient ISC process. The principal photophysical processes of the Pt(II) acetylide complexes are similar to that of the Ru(II) polyimine complexes, that is, the excitation into the 1MLCT excited state is followed by an efficient ISC to the triplet excited state, which was identified as 3MLCT/3LLCT transition. Two prominent photophysical features should be noted for Pt(II) acetylide complexes, i.e. the high phosphorescence quantum yield (up to 40%) and the readily tunable photophysical properties by simply changing the structure of the acetylide ligand.35–39In 2010, a trident Pt(II) acetylide complex, NˆNˆN Pt(II) phenylethynyl (Pt-3), was used for TTA based upconversion (Fig. 13).35 This complex shows moderate UV-vis absorption in the visible region (400 nm–550 nm), and a phosphorescence lifetime of 4.6 μs. The complex gives emission at 613 nm, as a broad emission band, due to the 3MLCT nature of the excited state. Accordingly DPA, a triplet acceptor with match energy level of T1 excited state (1.77 eV, 700 nm), was selected as the triplet acceptor/annihilator/emitter. With selective excitation at either 500 nm or 514.5 nm (laser), the upconverted blue emission of DPA was observed in the range of 400 nm–500 nm. The upconversion quantum yield (ΦUC) was determined as 0.2–1.1%.
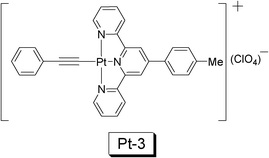 | ||
| Fig. 13 The NˆNˆN Pt(II) acetylide complex Pt-3 used for TTA upconversion. The upconversion with Pt-3 was reported in ref. 35. | ||
We propose that with NˆN Pt(II) acetylides complexes that show strong absorption and longer triplet excited state lifetimes, the upconversion quantum yield (ΦUC) may be greatly improved.
Following this line, recently we prepared a coumarin acetylide Pt(II) complex, which shows intense absorption in the visible region (Pt-4 in Fig. 14).36DPA was used as the triplet acceptor and upconversion quantum yield of 14.1% was observed.36 Under the same experimental conditions, the model complex dbbpyPt(II) bisphenylacetylide gives upconversion quantum yield of 8.9%. Herein we propose to use a new parameter to evaluate the upconversion performance of the triplet sensitizers (η, eqn 5). We compared the η value of the coumarin-containing complex Pt-4 and the model complex dbbpyPt(II) bisphenylacetylide, we found the overall upconversion capability with Pt-4 is improved by 7.3-fold over the model complex containing phenylacetylide.
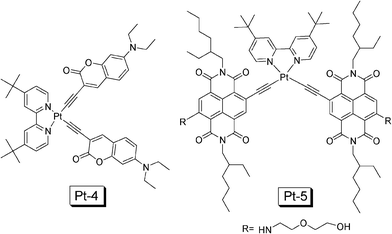 | ||
| Fig. 14 NˆNPt(II) complexes Pt-4 and Pt-5 that show intense absorption of visible light used as triplet sensitizers for TTA upconversion. The compounds Pt-4 and Pt-5 are from ref. 36 and 37, respectively. | ||
It should be noted that the absorption of Pt-4 is still at a short wavelength and the T1 excited state lifetime is short (2.5 μs).36 Therefore, recently we prepared complex Pt-5 (Fig. 14), in which the naphthalenediimide (NDI) was attached to the Pt(II) centre via an acetylide ligand.37 Intense absorption in the visible region (λabs = 583 nm with ε = 31300 M−1 cm−1) and long-lived T1 excited state was observed for the complex Pt-5 (τ = 22.3 μs), these photophysical properties are ideal for the complexes as triplet sensitizers for TTA upconversion. The upconversion quantum yield (ΦUC) of the complex was determined as 9.5%. Under the same experimental conditions, no upconversion was observed for the model complex dbbpyPt(II) bisphenylacetylide.
In 2010 we reported a naphthalimide (NI) acetylide-containing Pt(II) complex Pt-6 (Fig. 15), which shows an exceptionally long-lived 3IL excited state (τ = 124 μs).18,38 We also noticed another Pt(II) bisacetylide complex Pt-7 with a long-lived T1 excited state reported by Castellano et al.39 A long-lived T1 excited state of triplet sensitizer is beneficial for improvement of the efficiency of the TTET process, the critical process involved in the TTA upconversion. We observed exceptionally high upconversion quantum yields (ΦUC) of 39.9% for Pt-6 and 28.8% for Pt-7. To the best of knowledge, these values are the highest ever reported, most of the reported values are smaller than 20%.8
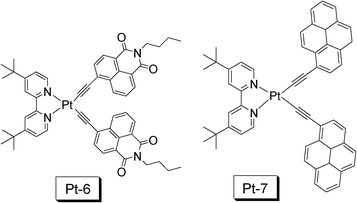 | ||
| Fig. 15 NˆNPt(II) bisacetylide complexes that show prolonged T1 excited state lifetimes (τ = 124.0 μs for Pt-6 and τ = 73.6 μs for Pt-7) used as triplet sensitizers for TTA upconversion. The compounds are from ref. 18 and 39, respectively. | ||
It should be pointed out that these high upconversion quantum yields are reasonable, although it was proposed that 11.1% will be the maximal upconversion quantum yields.
Recently we prepared a NˆNPt(II) acetylide complex with rhodamine moiety, in order to enhance the absorption in the visible region and to access the long-lived 3IL excited state localized on the rhodamine moiety (Pt-8. Fig. 16).40 The complex shows a strong absorption at 556 nm (ε = 185![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 M−1 cm−1). The difference between the complex and the model complex is substantial. By comparison the intense absorption of Pt-8 at 556 nm is due to the rhodamine acetylide ligand. Interestingly, only fluorescence (580 nm) was observed for Pt-8 and no phosphorescence was observed for Pt-8 at either RT or 77 K. The assignment of fluorescence is based on the small Stokes shift of the emission band (24 nm), short luminescence lifetime (2.50 ns) and its insensitivity to O2.40
800 M−1 cm−1). The difference between the complex and the model complex is substantial. By comparison the intense absorption of Pt-8 at 556 nm is due to the rhodamine acetylide ligand. Interestingly, only fluorescence (580 nm) was observed for Pt-8 and no phosphorescence was observed for Pt-8 at either RT or 77 K. The assignment of fluorescence is based on the small Stokes shift of the emission band (24 nm), short luminescence lifetime (2.50 ns) and its insensitivity to O2.40
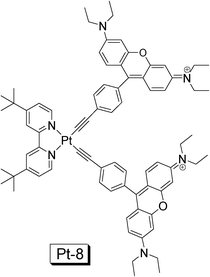 | ||
| Fig. 16 NˆNPt(II) acetylide complex containing rhodamine moiety. The complex shows strong absorption at 556 nm (ε = 185 800 M−1 cm−1) and long-lived non-emissive 3IL excited state was observed (τT = 83.0 μs). The complex is from ref. 40. | ||
Interestingly, nanosecond time-resolved transient difference absorption spectra show that rhodamine-localized triplet excited state was populated upon excitation of Pt-8. The lifetime of the triplet excited state is 83.0 μs. This assignment of the triplet excited state as 3IL state was supported by the position of the bleaching band and DFT calculations (spin density analysis of the triplet state of the complex).
The complex Pt-8 was used as the triplet sensitizer for TTA upconversion with perylene as the triplet acceptor and upconversion quantum yield of 11.2% was observed. Note the overall upconversion capability of Pt-8 is significant, due to its strong absorption at 556 nm (ε = 185800 M−1 cm−1).
2.4 Cyclometalated Pt(II)/Ir(III)complexes
Cyclometalated Ir(III) or Pt(II) complexes also show RT phosphorescence upon photoexcitation.41,42 The photophysical properties of these complexes, such as the absorption wavelength, the emission wavelength and to some extent, the lifetime of the T1 excited state, can be tuned by changing the CˆN ligand. Cyclometalated Ir(III) complexes are normally used as the triplet emitters in organic light emitting diodes (OLED), due to their triplet manifold of the emissive excited state.40 The emissive state of these complexes are generally assigned as 3MLCT/3IL mixed feature and the luminescence is characterized by a long lifetime (in the μs range). Therefore, these complexes can potentially be used as triplet sensitizers as the TTA based upconversion. However, it should be pointed out that the typical cyclometalated Ir(III) or Pt(II) complexes usually show weak absorption in the visible region and the lifetime of the T1 state is short (only a few μs).41–43In 2006, Castellano et al. used complex Ir-1 as the triplet sensitizer for the TTA based upconversion (Fig. 17).44 The lifetime of the triplet excited state of Ir-1 is 1.55 μs and the energy level of the T1 state is ca. 20000 cm−1 (500 nm, 2.48 eV). However, the absorption of this complex is weak in the visible region. Pyrene and di-(tert-butyl)pyrene were used as the triplet acceptor/annihilator, due to the appropriate energy level of the T1 excited state of pyrene or its derivative 3,8-di-tert-butylpyrene (16850 cm−1, i.e. 593 nm, 2.09 eV). Upconverted blue fluorescence of pyrene was observed at 400 nm with selective excitation of the triplet sensitizer at 450 nm.
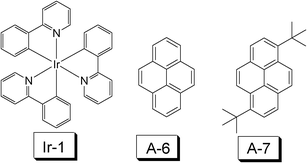 | ||
| Fig. 17 Molecular structures of the triplet sensitizer cyclometalated iridium complex Ir(ppy)3 (Ir-1, ppy = 4-phenylpyridine) (triplet sensitizer) and the triplet acceptor pyrene and 3,8-di-tert-butylpyrene (A-6 and A-7). The compounds are from ref. 44. | ||
However, it should be pointed out that the UV-vis absorption of complex Ir-1 is located in the UV and blue region, and the molar extinction coefficient is only moderate. Furthermore, the lifetime of the T1 excited state is short. Therefore, much room is left for the chemical modification of the molecular structure of the cyclometalated Ir(III) complexes to improve the UV-Vis absorption property and thus to enhance the TTA upconversion with these complexes.
Recently, inspired by Thompson's work,45a we prepared cyclometalated Ir(III) and Pt(II) complexes which contain a coumarin ligand (for example, Ir-3 and Ir-4, Fig. 18).45b,46 The design rationales are to prepare the Ir(III) and Pt(II) complexes that show intense absorption in the visible region and to access the long-lived 3IL excited state. With DFT calculations we predicted that the T1 energy level of the coumarin ligand will be close to the Ir(III) coordination center thus the intrinsic triplet excited state of the complex may be profoundly perturbed.
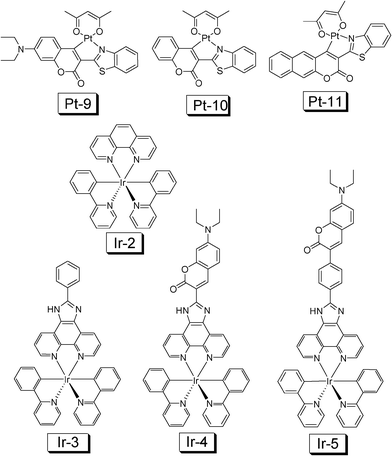 | ||
| Fig. 18 Cyclometalated Ir(III) and Pt(II) complexes used for TTA upconversion. Complexes Ir-4, Ir-5 and Pt-9 are with light-harvesting ligand. Note the Ir(III) complexes are cations. The complexes are from ref. 45 and 46. | ||
Upconversion with Pt-9 (τ = 20.3 μs) as the sensitizer and DPA as the acceptor gives an upconversion quantum yield of 15.4%. Upconversion with Pt-10 and Pt-11 give much lower efficiency.46
In contrast to the model complexes Ir-2 and Ir-3, both showing very weak absorption in the visible region (for example, ε = 1353 M−1 cm−1 at 466 nm for Ir-3), the coumarin-containing Ir-4 gives intense absorption (ε = 70920 M−1 cm−1 at 466 nm) (Fig. 19). Furthermore, the T1 excited state lifetimes of Ir-2 and Ir-3 are short (0.81 μs and 0.66 μs, respectively), Ir-4 shows a profoundly prolonged T1 excited state lifetime (τ = 75.5 μs).
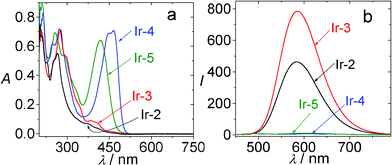 | ||
| Fig. 19 (a) UV-vis absorption of Ir-2, Ir-3, Ir-4 and Ir-5. In CH3CN (1.0 × 10−5 M; 20 °C). (b) Emission spectra of the IrIII complexes. Ir-2: λex = 386 nm, Ir-3: λex = 407 nm, Ir-4: λex = 462 nm, Ir-5: λex = 421 nm. In deaerated CH3CN (1.0 × 10−5 M; 20 °C). Reproduced with permission from ref. 45b. | ||
Interestingly, the coumarin-containing complexes Ir-4 and Ir-5 give much weaker emission than that of Ir-2 and Ir-3 (Fig. 19b).45b The emissive excited state of Ir-4 and Ir-5 were proposed to be 3IL excited state by using nanosecond time-resolved transient absorption, 77 K emission spectra and spin density analysis (DFT calculations). We propose that the weak emission of Ir-4 and Ir-5 do not necessarily deter the complexes from application for some photophysical process, thus the complexes were used for TTA based upconversion.
It is clear the Ir-4 and Ir-5 are more efficient as triplet sensitizers for TTA upconversion than the model complexes Ir-2 and Ir-3 (Fig. 20). For example, the upconversion quantum yields with Ir-4 and Ir-5 as triplet sensitizers were determined as 21.3% and 23.4%, respectively. For Ir-2 and Ir-3, however, no significant upconversion was observed (Fig. 20a).45b
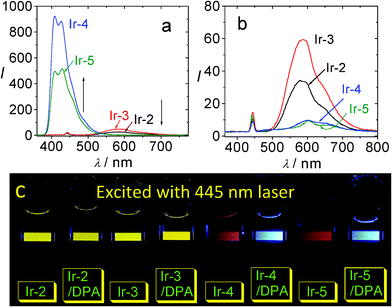 | ||
| Fig. 20 Upconversion with IrIII complexes as the triplet sensitizers and DPA as the triplet acceptor. (a) Upconversion emission spectra of the mixture of Ir-2, Ir-3, Ir-4 and Ir-5 (1.0 × 10−5 M) with DPA (8.0 × 10−5 M). (b) Phosphorescence of sensitizers alone (λex = 445 nm, 5 mW). (c) Photographs of the upconversions. In deaerated CH3CN. 20 °C. Reproduced with permission from ref. 45b. | ||
The upconversion are clearly visible with un-aided eyes. Herein we noticed an interesting result, i.e., the quenched phosphorescence peak areas of Ir-4 and Ir-5 are much smaller than that of the upconverted fluorescence peak area. This is abnormal since the traditional understanding of the TTA upconversion implies that the phosphorescence of the sensitizer will be quenched by triplet acceptors. The upconversion with Ir-4 and Ir-5 clearly show that some sensitizer molecules that are otherwise non-emissive were involved in the TTET process, that is, the dark excited states were effective as energy donors of the TTET process. It should be pointed out that previously all the transition metal complexes used as triplet sensitizers for TTA upconversion are phosphorescent. Our new concept to use dark triplet excited state to sensitize the TTET and the TTA upconversion will greatly increase the availability of the triplet sensitizers for TTA upconversion.45
The effect of the long-lived T1 excited state on the efficiency of the TTET process is presented in Fig. 21. The slope of the quenching process with Ir-4 and Ir-5, i.e. the quenching constants, are much larger than that with Ir-2 and Ir-3. The quenching constants of Ir-4 and Ir-5 are 50-fold of that with Ir-2 and Ir-3 as the triplet sensitizers.
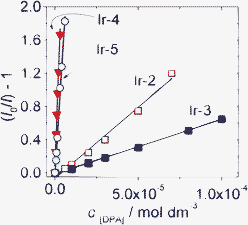 | ||
| Fig. 21 Stern–Volmer plots generated from phosphorescence intensity quenching of complex Ir-2 (λex = 386 nm), Ir-3 (λex = 410 nm), Ir-4 (λex = 475 nm) and Ir-5 (λex = 425 nm). Phosphorescence was measured as a function of DPA concentration in CH3CN. 1.0 × 10−5 mol dm−3. 20 °C. Reproduced with permission from ref. 45b. | ||
The Stern–Volmer quenching constants (KSV) of Ir-4 and Ir-5 with DPA as quencher were determined as 5.51 × 105 M−1 and 3.18 × 105 M−1, respectively. For Ir-2 and Ir-3, however, much smaller quenching constants of 1.71 × 104 M−1 and 6.57 × 103 M−1, were observed. Small Stern–Volmer quenching constants indicate a relatively non-efficient TTET process.
2.5 Organic triplet sensitizers
To date most of the triplet sensitizers used in TTA upconversion are transition metal complexes, due to the efficient ISC effect, thus population of the triplet excited state upon photoexcitation. However, these complexes are synthetically demanding and are expensive for applications. Similar to the development of the DSCs, for which a transition from metal complex sensitizers to organic sensitizers has been completed, neat organic sensitizers are desired for TTA upconversion. This is challenging because very few neat organic chromophores (metal-free) show efficient ISC effect and at the same time, show intense absorption of the visible light.However, some organic chromophores do show the Sm→Tn (m, n > 0) ISC without the need for any heavy atom effect, such as 2,3-butanedione, acridone and diphenyl ketone, etc. (Fig. 22). In these cases it is believed that the ISC is facilitated by the n–π* transitions. The energy gap between the S1 and T1 state is also important. For example, the diacetyl shows a room temperature phosphorescence quantum yield of 1.00. The room temperature phosphorescence lifetime is found to be with a 43 μs component (30%) and a shorter lived component of 11 μs (70%). Thus it is possible to use the triplet sensitizer for TTA based upconversion.472,5-Diphenyloxazole (PPO) was selected as the triplet acceptor (ΦF = 0.85) (A-8, Fig. 22).
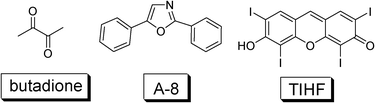 | ||
| Fig. 22 Molecular structures of organic triplet sensitizer 2,3-butanedione and TIHF (2,4,5,7-tetraiodo-6-hydroxy-3-fluorone).47,48 Triplet acceptor A-8 (2,5-diphenyloxazole) was used for upconversion with butadione. | ||
The upconverted blue fluorescence of A-8 in the range of 350–450 nm was observed with the selective excitation of the triplet sensitizer (2,3-butanedione) at 442 nm. The anti-Stokes shift is up to 0.64 eV.47
Besides transition metal atoms, the iodine atom also shows a moderate heavy atom effect, thus it is also possible that singlet→triplet transition can be facilitated with iodine. It should be pointed out that introduction of an iodine atom into the organic chromophores is relatively easy. Thus, the use of iodine-containing organic chromophores as triplet sensitizers for the TTA based upconversion is a promising substitute for the transition metal complex sensitizers.
Following this line, iodo-containing organic fluorophore 2,4,5,7-tetraiodo-6-hydroxy-3-fluorone (TIHF), was used as the triplet sensitizer for TTA based upconversion (Fig. 22).48 The dye shows a low fluorescence quantum yield (ΦF) of 0.13, but a higher quantum yield of the triplet excited state (ΦT = 0.87).49 In this case it was proved with experiments that the quantum yield of the triplet excited state is ΦT = 1 − ΦF. It should be pointed out that the quantum yield of the triplet excited state is not necessarily always ΦT = 1 − ΦF.
Dye
TIHF shows ideal properties for TTA based upconversion: intense absorption in the visible region (ε = 91200 M−1 cm−1 at 536 nm) and a long triplet excited state lifetime (τT = 25 μs, in ethanol).49DPA was used as the triplet acceptor/annihilator. The upconversion quantum yield is 0.6%.
It should be pointed out that limitations still exist for this system. First, it is difficult to change the molecular structure of TIHF to tune the energy level of the triplet excited state (T1) and the singlet excited state (UV-vis absorption wavelength). Second, the lifetime of the triplet excited state of TIHF is short for the triplet excited state of an organic chromophore.
Along this line, an organic chromophore with lower fluorescence quantum yield, long-lived triplet excited state, strong absorption in the visible region will be greatly desired for the TTA based upconversion. As we pointed out earlier, the scaffold of TIHF is not a general platform because the molecular structure can not be readily modified, as a result, the photophysical properties concerning the application in upconversion can not be readily tuned.
In order to address this challenge, recently we devised a small library of organic triplet sensitizers from a single chromophore of BODIPY (Fig. 23, B-1–B-7).50 The absorption of the sensitizers cover a wide range of 510–629 nm, the variation of the absorption wavelength of the sensitizers is achieved by extension of the π-conjugation framework of the molecules. The molar extinction coefficients (ε) of the sensitizers are up to 180000 M−1 cm−1. These triplet sensitizers give weak fluorescence. The lifetimes of the triplet excited states of the sensitizers are up to 66.3 μs. DFT calculations predict that the T1 energy levels of these sensitizers are at 800–900 nm range. B-3 gives a much lower T1 energy level (1075 nm) than the other sensitizers. With perylene (Fig. 9) or 1-chloro-9,10-bis(phenylethynyl) anthracene (1CBPEA, Fig. 23) as the triplet acceptors, significant upconversion (ΦUC up to 6.1%) was observed for solution samples and polymer films, and the anti-Stokes shift was up to 0.56 eV.50 We attribute the efficient upconversion with B-1–B-7 to the iodo substitutions, which are in direct connection with the BODIPY core. A model compound B-8 was also studied as the triplet sensitizer, but no upconversion was observed, which is due to the large distance between the iodo substitution and the BODIPY core, thus the weak heavy atom effect will lead to a poor ISC effect. Note that the phenyl group in B-8 is not in the π-conjugation framework.
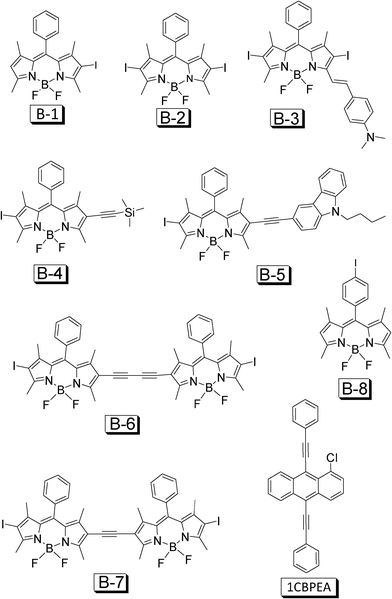 | ||
| Fig. 23 BODIPY based organic triplet sensitizers for TTA upconversion. The triplet acceptor 1-chloro-bis-phenyl ethynylanthracene (1CBPEA) is also presented. The compounds are from ref. 50. | ||
We believe that the devise of B-1–B-7 (Fig. 23) is only the beginning of the development of organic triplet sensitizers for TTA upconversion. Considering the great availability of organic chromophores, much room is left for development of neat organic triplet sensitizers for TTA upconversion.
2.6 Sensitizers with non-emissive T1 excited states
Inspired by the organic triplet sensitizer, for which the T1 state is usually non-emissive,47,48,50 such as B-1–B-7 (Fig. 23), we envisaged that non-phosphorescent transition metal complexes with triplet excited states populated upon photoexcitation can sensitize the TTA upconversion. Recently, we reported a Ru(II) polyimine-coumarin dyad that shows a non-emissive 3IL exited state and gives very weak phosphorescence but significant upconversion capability (Ru-10. Fig. 24).51 We propose that the phosphorescence is actually detrimental to the TTET process as well as upconversion because the radiative decay of the triplet excited state of the sensitizer (i.e., phosphorescence) is competitive to TTET.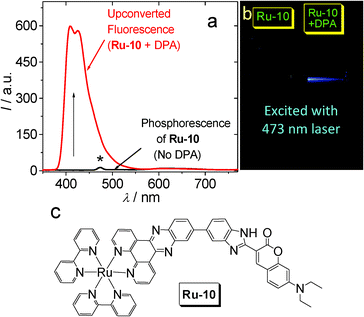 | ||
| Fig. 24 (a) Upconversion with non-emissive Ru-10 as the triplet sensitizer and DPA as the triplet acceptor. Excited by 473 nm laser. The asterisk indicates laser scattering. (b) Photographs of the upconversion (samples from a). (c) Molecular structure of triplet sensitizer Ru-10. Adapted with permission from ref. 51. | ||
3. Triplet acceptors of TTA upconversion
Compared to the development of triplet sensitizers for TTA upconversion, much less attention has been paid to the development of triplet acceptors. To date the triplet acceptors are limited to the commercially available compounds, very few are with tailored design.Based on the reported TTA upconversion examples (Fig. 25), we can summarize the common requirement for the triplet acceptors. (1) The energy level should follow the relation of 2 × ET1 > ES1, thus the annihilation of the triplet excited state can produce the singlet excited state. (2) High fluorescence quantum yield, if photon emission is desired for the TTA upconversion. (3) Tunable T1 excited state energy level. (4) Good photochemical stability.
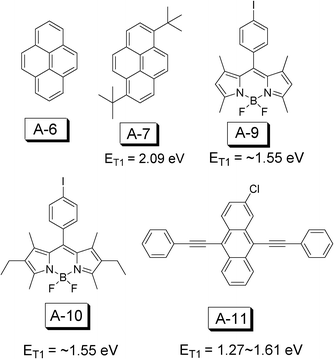 | ||
| Fig. 25 Molecular structures of triplet acceptors A-6, A-7, A-9, A-10 and A-11 for TTA upconversion. | ||
The triplet acceptors used for TTA upconversion are anthracene, 9,10-diphenylanthracene (DPA), perylene, and boron-dipyrromethene (BODIPY) etc. (Fig. 25).
2-Chloro-bis-phenylethy phenylethynylanthracene (A-11, 2CBPEA) was used as a triplet acceptor with red absorbing platinum(II) tetraphenyltetrabenzoporphyrin (PtTPBP) as the sensitizer.52 The T1 excited state of A-11 was estimated to be between 1.27–1.61 eV. A-11 gives emission in the range of 475–625 nm, with emission bands centered at 480 nm and 515 nm.52
BODIPY dye A-10 (Fig. 25) was used as the triplet acceptor, which shows a T1 excited state energy level at ca. 800 nm. The red light absorbing platinum(II) tetraphenyltetrabenzoporphyrin (PtTPBP) was used as the triplet sensitizer (Φp = 0.7, τ = 40.6 μs in benzene). Upconversion quantum yield (ΦUC) up to 15% was observed with excitation at 635 nm, where the I-BODIPYs give no absorption.53
4. Conclusions and outlook
Triplet–triplet annihilation (TTA) based upconversion is a promising upconversion scheme due to the low excitation power requirement (a few mW cm−2 is sufficient, unfocused terrestrial solar irradiance is 100 mW cm−2), readily tunable excitation/emission wavelength, intense absorption of the excitation light and high upconversion quantum yields. These advantages over other upconversion approaches are ideal for applications such as in photovoltaics, photocatalysis, etc. Furthermore, it has been demonstrated that the upconversion is effective in solid matrixes, such as in polymer films or dendrimers.29,54,55 The success of this observation of TTA upconversion in a solid matrix is probably due to the large Dexter distance, for example, up to 26.5 Å with PtOEP as the sensitizer and DPA as the acceptor was reported.11 However, much room is left for the development of the TTA upconversion. For example, the current triplet sensitizers are limited to the off-the-shelf transition metal complexes, tailored design sensitizers or neat organic triplet sensitizers are rarely reported. New sensitizers with intense absorption of visible light, especially in the red and near-IR range, and long-lived T1 excited state are highly desired. Herein we propose a new parameter, the overall upconversion capability η = ε × ΦUC, to evaluate the overall upconversion capability of the TTA upconversion, especially for applications of TTA upconversion, where ε is the molar extinction coefficient of the sensitizer at the excitation wavelength and ΦUC is the upconversion quantum yield. Second, little attention has been paid to the development of triplet acceptors, which is also important for the TTA upconversion. We believe that chemists will play a critical role in the development of TTA upconversion, because design of the triplet sensitizers and acceptors are dependent on molecular engineering. However, photophysics has to be considered in the molecular design because energy levels of the singlet and triplet excited states of the sensitizers and acceptors must follow some rules (see the Jablonski diagram in Scheme 1). TTA upconversion is probably the most promising upconversion approach and will flourish in the coming years.Acknowledgements
We thank the NSFC (20972024 and 21073028), the Royal Society (UK) and NSFC (China-UK Cost-Share Science Networks, 21011130154), the Fundamental Research Funds for the Central Universities (DUT10ZD212 and DUT11LK19) and Ministry of Education (SRFDP-200801410004 and NCET-08-0077) for financial support. Last but not least, we are grateful to all the colleagues around the world for their enthusiastic contributions to this fascinating research area.References
- C. Reinhard, R. Valiente and H. U. Guldel, J. Phys. Chem. B, 2002, 106, 10051 CrossRef CAS.
- M. Haase and H. Schafer, Angew. Chem., Int. Ed., 2011, 50, 5808 CrossRef CAS.
- I. Etchart, M. Berard, M. Laroche, A. Huignard, I. Hernandez, W. P. Gillin, R. J. Curryd and A. K. Cheetham, Chem. Commun., 2011, 47, 6263 RSC.
- W. Niu, S. Wu, S. Zhang and L. Li, Chem. Commun., 2010, 46, 3908 RSC.
- S. Baluschev, T. Miteva, V. Yakutkin, G. Nelles, A. Yasuda and G. Wegner, Phys. Rev. Lett., 2006, 97, 143903 CrossRef CAS.
- S. Baluschev, V. Yakutkin, T. Miteva, G. Wegner, T. Roberts, G. Nelles, A. Yasuda, S. Chernov, S. Aleshchenkov and A. Cheprakov, New J. Phys., 2008, 10, 013007 CrossRef.
- A. Monguzzi, J. Mezyk, F. Scotognella, R. Tubino and F. Meinar, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 195112 CrossRef.
- T. N. Singh-Rachford and F. N. Castellano, Coord. Chem. Rev., 2010, 254, 2560 CrossRef CAS.
- Y. Y. Cheng, T. Khoury, R. G. C. R. Clady, M. J. Y. Tayebjee, N. J. Ekins-Daukes, M. J. Crossley and T. W. Schmidt, Phys. Chem. Chem. Phys., 2010, 12, 66 RSC.
- T. N. Singh-Rachford, R. R. Islangulov and F. N. Castellano, J. Phys. Chem. A, 2008, 112, 3906 CrossRef CAS.
- A. Monguzzi, R. Tubino and F. Meinardi, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 155122 CrossRef.
- J. Saltiel, G. R. Marchand, W. K. Smothers, S. A. Stout and J. L. Charlton, J. Am. Chem. Soc., 1981, 103, 7159 CrossRef CAS.
- P. P. Levin, Dokl. Phys. Chem., 2003, 388, 10 CrossRef CAS.
- R. R. Islangulov, D. V. Kozlov and F. N. Castellano, Chem. Commun., 2005, 3776 RSC.
- D. V. Kozlov and F. N. Castellano, Chem. Commun., 2004, 2860 RSC.
- S. Ji, W. Wu, W. Wu, P. Song, K. Han, Z. Wang, S. Liu, H. Guo and J. Zhao, J. Mater. Chem., 2010, 20, 1953 RSC.
- W. Wu, W. Wu, S. Ji, H. Guo, P. Song, K. Han, L. Chi, J. Shao and J. Zhao, J. Mater. Chem., 2010, 20, 9775 RSC.
- H. Guo, S. Ji, W. Wu, W. Wu, J. Shao and J. Zhao, Analyst, 2010, 135, 2832 RSC.
- J. R. Winkler, T. L. Netzel, C. Creutz and N. Sutin, J. Am. Chem. Soc., 1987, 109, 2381 CrossRef CAS.
- E. A. Medlycott and G. S. Hanan, Chem. Soc. Rev., 2005, 34, 133 RSC.
- M. Abrahamsson, M. Jager, R. J. Kumar, T. Osterman, P. Persson, H. –C. Becker, O. Johansson and L. Hammarstrom, J. Am. Chem. Soc., 2008, 130, 15533 CrossRef CAS.
- X.-Y. Wang, A. D. Guerzo and R. H. Schmehl, J. Photochem. Photobiol., C, 2004, 5, 55 CrossRef CAS.
- N. D. McClenaghan, Y. Leydet, B. Maubert, M. T. Indelli and S. Campagna, Coord. Chem. Rev., 2005, 249, 1336 CrossRef CAS.
- A. I. Baba, J. R. Shaw, J. A. Simon, R. P. Thummel and R. H. Schmehl, Coord. Chem. Rev., 1998, 171, 43 CrossRef CAS.
- N. Armaroli, ChemPhysChem, 2008, 9, 371 CrossRef CAS.
- S. Ji, W. Wu, W. Wu, H. Guo and J. Zhao, Angew. Chem., Int. Ed., 2011, 50, 1626 CrossRef CAS.
- K. Kalyanasundaram, Photochemistry of Polypyridine and Porphyrin Complexes, Academic Press, New York, 1992, p. 500 Search PubMed.
- D. B. Papkovsky and T. C. O'Riordan, J. Fluoresc., 2005, 15, 569 CrossRef CAS.
- R. R. Islangulov, J. Lott, C. Weder and F. N. Castellano, J. Am. Chem. Soc., 2007, 129, 12652 CrossRef CAS.
- T. N. Singh-Rachford and F. N. Castellano, J. Phys. Chem. Lett., 2010, 1, 195 Search PubMed.
- S. Baluschev, V. Yakutkin, T. Miteva, Y. Avlasevich, S. Chernov, S. Aleshchenkov, G. Nelles, A. Cheprakov, A. Yasuda, K. Müllen and G. Wegner, Angew. Chem., Int. Ed., 2007, 46, 7693 CrossRef CAS.
- S. Baluschev, V. Yakutkin, G. Wegner, T. Miteva, G. Nelles, A. Yasuda, S. Chernov, S. Aleshchenkov and A. Cheprakov, Appl. Phys. Lett., 2007, 90, 181103 CrossRef.
- P. E. Keivanidis, S. Baluschev, T. Miteva, G. Nelles, U. Scherf, A. Yasuda and G. Wegner, Adv. Mater., 2003, 15, 2095 CrossRef CAS.
- T. N. Singh-Rachford, A. Nayak, M. L. Muro-Small, S. Goeb, M. J. Therien and F. N. Castellano, J. Am. Chem. Soc., 2010, 132, 14203 CrossRef CAS.
- P. Du and R. Eisenberg, Chem. Sci., 2010, 1, 502 RSC.
- H. Sun, H. Guo, W. Wu, X. Liu and J. Zhao, Dalton Trans., 2011, 40, 7834 RSC.
- Y. Liu, W. Wu, J. Zhao, X. Zhang and H. Guo, Dalton Trans., 2011, 40, 9085 RSC.
- H. Guo, M. L. Muro-Small, S. Ji, J. Zhao and F. N. Castellano, Inorg. Chem., 2010, 49, 6802 CrossRef CAS.
- I. E. Pomestchenko, C. R. Luman, M. Hissler, R. Ziessel and F. N. Castellano, Inorg. Chem., 2003, 42, 1394 CrossRef CAS.
- L. Huang, L. Zeng, H. Guo, W. Wu, W. Wu, S. Ji and J. Zhao, Eur. J. Inorg. Chem., 2011 DOI:10.1002/ejic.201100777.
- L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigellet, Top. Curr. Chem., 2007, 281, 143 CAS.
- J. A. Gareth Williams, Top. Curr. Chem., 2007, 281, 205 CAS.
- K. Hanson, A. Tamayo, V. V. Diev, M. T. Whited, P. I. Djurovich and M. E. Thompson, Inorg. Chem., 2010, 49, 6077 CrossRef CAS.
- W. Zhao and F. N. Castellano, J. Phys. Chem. A, 2006, 110, 11440 CrossRef CAS.
- (a) J. Brooks, Y. Babayan, S. Lamansky, P. I. Djurovich, I. Tsyba, R. Bau and M. E. Thompson, Inorg. Chem., 2002, 41, 3055 CrossRef CAS; (b) J. Sun, W. Wu, H. Guo and J. Zhao, Eur. J. Inorg. Chem., 2011, 3165 CrossRef CAS.
- W. Wu, W. Wu, S. Ji, H. Guo and J. Zhao, Dalton Trans., 2011, 40, 5953 RSC.
- T. N. Singh-Rachford and F. N. Castellano, J. Phys. Chem. A, 2009, 113, 5912 CrossRef CAS.
- H.–C. Chen, C.–Y. Hung, K.–H. Wang, H.–L. Chen, W. S. Fann, F.-C. Chien, P. Chen, T. J. Chow, C.–P. Hsu and S.–S. Sun, Chem. Commun., 2009, 4064 RSC.
- E. Klimtchuk, M. A. J. Rodgers and D. C. Neckem, J. Phys. Chem., 1992, 96, 9817 CrossRef CAS.
- W. Wu, H. Guo, W. Wu, S. Ji and J. Zhao, J. Org. Chem., 2011, 76, 7056 CrossRef CAS.
- S. Ji, H. Guo, W. Wu, W. Wu and J. Zhao, Angew. Chem., Int. Ed., 2011, 50, 8283 CAS.
- T. N. Singh-Rachford and F. N. Castellano, Inorg. Chem., 2009, 48, 2541 CrossRef CAS.
- T. N. Singh-Rachford, A. Haefele, R. Ziessel and F. N. Castellano, J. Am. Chem. Soc., 2008, 130, 16164 CrossRef CAS.
- K. Tanaka, K. Inafuku and Y. Chujo, Chem. Commun., 2010, 46, 4378 RSC.
- P. B. Merkel and J. P. Dinnocenzo, J. Lumin., 2009, 129, 303 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |