Anion templated assembly of [2]catenanes capable of chloride anion recognition in aqueous solvent media†
Nicholas H.
Evans
a,
Emma S. H.
Allinson
a,
Michael D.
Lankshear
a,
Ka-Yuen
Ng
a,
Andrew R.
Cowley
a,
Christopher J.
Serpell
a,
Sérgio M.
Santos
b,
Paulo J.
Costa
b,
Vítor
Félix
b and
Paul D.
Beer
*a
aInorganic Chemistry Laboratory, Department of Chemistry, University of Oxford, South Parks Road, Oxford, OX1 3QR, UK. E-mail: paul.beer@chem.ox.ac.uk
bDepartamento de Química, CICECO and Secção Autónoma de Ciências da Saúde, Universidade de Aveiro, 3810-193, Aveiro, Portugal
First published on 28th September 2011
Abstract
An anion templated double cyclization strategy to synthesize [2]catenanes in which two identical acyclic pyridinium receptor motifs interweave around a chloride anion template is described. Ring closing metathesis (RCM) of the preorganized orthogonal precursor chloride complex facilitates the isolation of [2]catenanes in very high yields. X-ray crystal structures provide an insight of the supramolecular forces responsible for chloride anion templated efficacy and recognition. Removal of the chloride anion template generates topologically unique interlocked binding cavities for anions. 1H NMR anion binding investigations demonstrate the catenanes to be highly selective hosts for chloride in preference to more basic monocharged oxoanions. In aqueous solvent media containing 30% water, such catenanes exclusively bind chloride, under which conditions no binding of acetate or dihydrogen phosphate is observed. Molecular dynamic simulations in the solution phase are used to account for the catenanes’ anion recognition properties.
Introduction
Since their initial postulation, interpenetrated and interlocked molecular architectures have provided a perpetual challenge to the inquisitive chemist. Driven by potential applications in, for example nanomachinery1 but also in no small part by aesthetic appeal, the field has evolved to be distinct in its own right.2 Given the complexity of these molecules, their syntheses are non-trivial. Since the initial statistical approaches,3 a range of cationic and neutral template methods have enabled a large catalogue of mechanically bonded systems to be constructed.4,5 However, the use of anion templates to synthesize interlocked molecules is underdeveloped, which has been attributed to the problems encountered in the binding of anions: namely, their small charge/radius ratio compared to cations, pH dependence, high solvation energies, weak coordination preferences (e.g. lack of any ligand field effects) and range of geometries.6 Inspired by the work of Sauvage,4a we set ourselves the challenge of exploiting anions to template the formation of interlocked supramolecular assemblies. To this end we have shown that pseudorotaxane,7 rotaxane8 and catenane9 formation can be templated selectively by a chloride anion which facilitates the interpenetration of a pyridinium threading component through the annulus of an isophthalamide macrocycle.10We are now able to demonstrate in a directly analogous approach to the pioneering work of Sauvage using the copper(I) cation template, that two identical acyclic pyridinium components are capable of associating around a single chloride anion in an orthogonal assembly, whereupon double cyclization affords [2]catenanes in very high yields (Scheme 1). X-ray crystal structures of the [2]catenanes confirm their interlocked nature and encapsulation of chloride within the catenanes’ topologically unique binding cavity. Upon removal of the chloride anion template, the [2]catenanes display strong binding for chloride (in up to 30% water) as compared to more basic, larger singly charged oxoanions. Molecular dynamic simulations rationalise the observed selectivity trends by revealing the catenane binding domains are of complementary size to the chloride anion, whereas the complexation of larger polyatomic anions such as acetate and dihydrogen phosphate results in unfavourable distortions of the interlocked cavities.11
![Schematic representation of the chloride anion templated double cyclization strategy for the synthesis of [2]catenanes.](/image/article/2011/RA/c1ra00394a/c1ra00394a-s1.gif) | ||
| Scheme 1 Schematic representation of the chloride anion templated double cyclization strategy for the synthesis of [2]catenanes. | ||
Precursor design and synthesis
Three precursor compounds 1+(X−), 2+(X−) and 3+(X−) (Fig. 1) were designed to incorporate complementary supramolecular interactions which would assist the molecule to assemble around a chloride anion template in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric host to guest fashion.
:1 stoichiometric host to guest fashion.
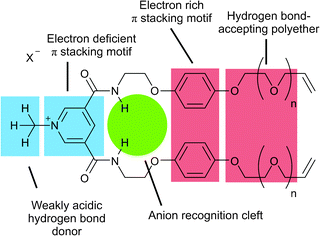 | ||
| Fig. 1 Structural design of precursor 1+(X−) (n = 2), 2+(X−) (n = 1) and 3+(X−) (n = 0). | ||
Each acyclic precursor molecule provides two amide hydrogen bond donating groups for anion binding such that a pseudo-tetrahedral amide hydrogen bonding association of two molecules of, for example, 1+ can occur around a spherical chloride anion in an orthogonal fashion. In addition, the electron deficient pyridinium motif was designed to intercalate between the electron rich hydroquinone rings of the other precursor in the 2:1 associated assembly. Further stabilization of the assembly could be provided by hydrogen bonding between the weakly acidic pyridinium N+-CH3 protons of one precursor molecule and the polyether oxygen atoms of the other. With this in mind the polyether chain length attached to the vinyl functional groups was varied in an effort to optimize the yield of [2]catenane formation of the ring closing metathesis (RCM) double cyclization reaction.
Compounds 1+(X−), 2+(X−) and 3+(X−) (X− = Cl−, PF6−) were prepared via the condensation of two equivalents of the corresponding vinyl appended amine compounds 4, 5 and 6 with 3,5-bis(chlorocarbonyl)-pyridine in dry CH2Cl2 in the presence of triethylamine to give the bis-amides 7, 8, 9 respectively. Methylation gave the pyridinium iodide compounds 1+(I−), 2+(I−) and 3+(I−). Subsequent anion exchanges afforded the chloride and hexafluorophosphate salts (Scheme 2).
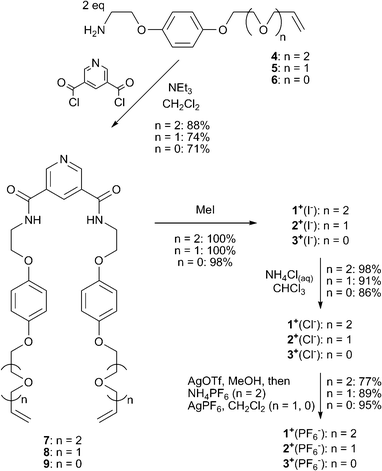 | ||
| Scheme 2 Synthesis of RCM precursors 1+(X−), 2+(X−) and 3+(X−) (X− = Cl−, PF6−). | ||
Catenane synthesis—importance of chloride anion template and Grubbs’ catalyst
Initially, the RCM reactions of acyclic precursors 1+(Cl−) and 1+(PF6−) were investigated.11 These were simply carried out by adding the precursors to CH2Cl2, followed by 10% (by wt) Grubbs’ 1st generation catalyst (Grubbs’ I) (Scheme 3).![Synthesis of [2]catenanes 102+(X−)(Y−).](/image/article/2011/RA/c1ra00394a/c1ra00394a-s3.gif) | ||
| Scheme 3 Synthesis of [2]catenanes 102+(X−)(Y−). | ||
Cyclization of precursor 1+(Cl−), produced [2]catenane 102+(Cl−)2 in an isolated yield of 34%, whereas an equimolar mixture of 1+(Cl−) and 1+(PF6−) afforded [2]catenane 102+(Cl−)(PF6−) in a yield of 78%. The significant increase in catenane yield in the latter case was attributed to the removal of the excess amount of chloride anion template, which introduced competition from a 1:1 binding mode between 1+ and the chloride anion that disfavours the formation of the [2]catenane structure.12 RCM of 1+(PF6−) afforded 102+(PF6−)2 in only 16% yield which indicates that π–π stacking alone does result in catenane formation, if only in modest yields.
More thorough investigations were undertaken using acyclic precursors 2+(Cl−) and 2+(PF6−) (Scheme 4): particular attention was focused on the generation of Grubbs’ catalyst (I or II) used. Yields of formation of catenanes 112+(X−)(Y−) are summarised in Table 1. At first appearance, broadly similar trends are observed in the yields of catenane formation, however, there are notable differences. Firstly, there is the formation of two catenane species when an equimolar mixture of 2+(Cl−) and 2+(PF6−) are cyclized, in a very high combined yield. One of these is the dichloride salt 112+(Cl−)2, identified by the 1H NMR spectrum matching that of the catenane species isolated from the cyclization of 2+(Cl−), as well as the lack of any peaks in the 19F NMR spectrum of the species. The other catenane species is assigned to be the mixed salt 112+(Cl−)(PF6−).13 The remarkably high combined yields of catenane formation (95% to quantitative, based on the consumption of chloride precursor 2+(Cl−)) are very impressive considering the double cyclization event. There is also a surprisingly high yield for the formation of 112+(PF6−)2 from cyclizing 2+(PF6−) with Grubbs’ I catalyst. This result may be attributed to the combination of subtle differences in reactivity of the type of Grubbs’ catalyst used14 and the different length of polyether chain which significantly increases the favourable RCM reaction over other metathesis reaction pathways.
![Synthesis of [2]catenanes 112+(X−)(Y−)](/image/article/2011/RA/c1ra00394a/c1ra00394a-s4.gif) | ||
| Scheme 4 Synthesis of [2]catenanes 112+(X−)(Y−) | ||
Small scale RCM test-reactions on precursor compounds 3+Cl− and 3+PF6− failed to produce any [2]catenane product. It was found that hexafluorophosphate salt 3+(PF6−) was insoluble in CH2Cl2, with no RCM being observed upon addition of Grubbs’ I catalyst to a suspension of 3+(PF6−) in dry CH2Cl2. Chloride salt 3+(Cl−) did cyclize but only to form macrocycle 13+(Cl−) (as verified by an isotope pattern of Δm/z = 1.0 in the ESMS), with both Grubbs’ I and II catalysts (Scheme 5).
![Unsuccessful preparation of [2]catenane 122+(Cl−)2 and formation of macrocycle 13+(Cl−).](/image/article/2011/RA/c1ra00394a/c1ra00394a-s5.gif) | ||
| Scheme 5 Unsuccessful preparation of [2]catenane 122+(Cl−)2 and formation of macrocycle 13+(Cl−). | ||
To investigate the origin of this failure to produce any [2]catenanes 122+(X−)(Y−), new macrocycle 14 was prepared (see ESI†). The addition of one equivalent of pyridinium hexyl chloride thread 15+(Cl−)7 to a CD2Cl2 solution of macrocycle 14 revealed no significant shifts in the 1H NMR spectrum indicating that no pseudorotaxane was being formed (Scheme 6 and Fig. S12 in ESI†). This experiment indicates the cavity of 14 is too small for the threading of a pyridinium unit, which implies the target catenane cannot be formed by cyclization of precursors 3+(Cl−) and 3+(PF6−).
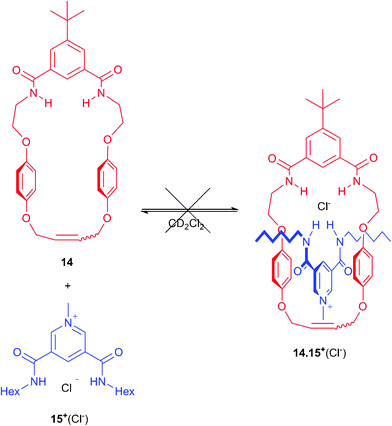 | ||
| Scheme 6 Failed pseudorotaxane formation between macrocycle 14 and thread 15+(Cl−). | ||
Characterization of [2]catenanes—evidence of interlocked nature
The interlocked nature and presence of various supramolecular interactions in the [2]catenane species 112+(X−)(Y−) are exemplified by the comparison of the 1H NMR spectra of catenane 112+(Cl−)2 to that of precursor 2+(Cl−) (Fig. 2). Upon catenation amide proton d and aromatic pyridinium proton c are notably more upfield, reflecting the change in ratio of pyridinium motif to chloride bound in cleft from 1:1 in the precursor to 2:1 in the catenane. There is also a large upfield shift and splitting of the hydroquinone protons g and h which is diagnostic of π–π stacking between the electron rich hydroquinone groups and electron deficient pyridinium motif. Finally, a modest downfield shift in the N-methyl pyridinium resonance a is observed, indicating the presence of hydrogen bonding of these protons to the polyether oxygen atoms.11
![1H NMR spectra of (a) RCM precursor 2+(Cl−) and (b) [2]catenane 112+(Cl−)2. Solvent: CD3CN. T = 293 K.](/image/article/2011/RA/c1ra00394a/c1ra00394a-f2.gif) | ||
| Fig. 2 1H NMR spectra of (a) RCM precursor 2+(Cl−) and (b) [2]catenane 112+(Cl−)2. Solvent: CD3CN. T = 293 K. | ||
Further evidence of the interlocked nature of [2]catenane 112+(X−)(Y−) was confirmed through variable cone voltage electrospray mass spectrometric (ESMS) experiments. At low cone voltage both the catenane dication and the singly charged chloride complex are observed in ESMS (Fig. 3a). However, considering that a [1 + 1] macrocycle could produce the same molecular ion signals, spectra of the [2]catenanes were recorded again at increased cone voltage to obtain unambiguous identification of the interlocked structure. Cleavage of one of the interlocked rings in a catenane would leave the other intact, producing a distinctive MS signal of the remaining macrocycle. The [1 + 1] macrocycle, however, is expected to fragment into a collection of random lower mass species. The observation of an intense peak, corresponding to the singly charged macrocycle, at m/z = 592.3 with Δm/z = 1.0 in the variable cone voltage ESMS experiment, confirms the interlocked nature of the double cyclization product (Fig. 3b).
![Electrospray mass spectra of [2]catenane 112+(Cl−)2 with cone voltage: (a) 30 V and (b) 80 V.](/image/article/2011/RA/c1ra00394a/c1ra00394a-f3.gif) | ||
| Fig. 3 Electrospray mass spectra of [2]catenane 112+(Cl−)2 with cone voltage: (a) 30 V and (b) 80 V. | ||
Yellow crystals of [2]catenane 112+(Cl−)2 suitable for single crystal X-ray structural analysis were grown by diffusion of di-isopropyl ether into a CHCl3/CH3OH solution of the catenane. The structure (Fig. 4) confirms the interlocked nature of the catenane, and that only one chloride anion may fit within the interlocked cavity of the molecule. The encapsulated chloride is held in place by six hydrogen bonds: four amide N–H⋯Cl− (N to Cl distances: 3.350 Å, 3.399 Å, 3.404 Å, 3.502 Å) and two para-pyridinium C–H⋯Cl− (C to Cl distances: 3.427 Å and 3.442 Å). There is evidence of some hydrogen bonding between the N-methyl pyridinium protons and the polyether oxygen atoms: one methyl group has two possible hydrogen bonds with C to O distances of 3.462 Å and 3.514 Å, while the other has a single C to O distance (below 4 Å) of 3.522 Å. The face-to-face stacking of the pyridinium ring and hydroquinone units is considerably offset from ideality: this is attributed to the steric demands of the spacer between the anion binding unit and the hydroquinones, the length and flexibility of the polyether chains, and the secondary hydrogen bonding between these chains and the pyridinium N-methyl groups.
![The crystal structure of [2]catenane 112+(Cl−)2. Hydrogen atoms (except for those involved in hydrogen bonding to the chloride anion) have been omitted.](/image/article/2011/RA/c1ra00394a/c1ra00394a-f4.gif) | ||
| Fig. 4 The crystal structure of [2]catenane 112+(Cl−)2. Hydrogen atoms (except for those involved in hydrogen bonding to the chloride anion) have been omitted. | ||
Anion binding studies
Investigations into the anion binding properties of [2]catenane 112+(PF6−)2 were undertaken by 1H NMR titration experiments. Aliquots of tetrabutylammonium (TBA) salts of chloride, acetate and dihydrogenphosphate were added to NMR samples of the catenane in 1:1 CDCl3:CD3OD. The anion binding events were observed to be fast on the NMR timescale, allowing for the calculation of association constants by the computer program EQNMR,15 with titration data obtained by monitoring the ortho-pyridinium signal fitting to a 1:1 binding model (see Fig. 5a and Table 2). The topologically unique cavity of catenane 112+(PF6−)2 confers a very significant increase in both the binding strength and selectivity for chloride to the interlocked species in comparison to acyclic receptor 2+(PF6−).9a,16 The impressive selectivity for chloride over acetate was also observed for catenane 102+(PF6−)2 in analogous 1H NMR titration experiments carried out in 1:1 CDCl3:CD3C(O)CD3.11 These observations can be rationalised by the fact that only chloride is bound and encapsulated within the interlocked cavity, whereas the oxoanions are too large to penetrate the cavity, and associate peripherally instead. Evidence for this theory may be found in the appearance of the titration curves associated with the para-pyridinium proton (Fig. 5b): only for chloride does the signal move downfield for the entire titration, whereas for H2PO4− and AcO− after an initial modest downfield shift, there is a subsequent upfield shift inferring an alternative mode of binding for these two anions.
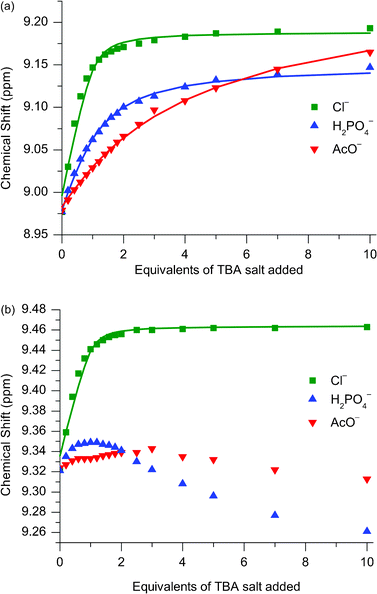 | ||
| Fig. 5
1H NMR titration data for catenane 112+(PF6−)2 against TBA salts of Cl−, H2PO4− and AcO− monitoring (a) ortho-pyridinium proton and (b) para-pyridinium proton. Solvent: 1:1 CDCl3:CD3OD. T = 293 K. | ||
| Cl− | H2PO4− | AcO− | |
|---|---|---|---|
|
a
T = 293 K. Error of experimental data fitting to calculated binding isotherm < 10%.
b Solvent: 1:1 CDCl3:CD3OD.
c Solvent: 1:1 CDCl3:CD3C(O)CD3.
d Data could not be fitted.
|
|||
| 2+(PF6−)b | K 11 = 230 | K 11 = 1360 K12 = 370 | K 11 = 1500 K12 = 345 |
| 112+(PF6−)2b | K 11 > 104 | K 11 = 1240 | K 11 = 160 |
| 102+(PF6−)2c | K 11 = 9240 K12 = 160 | —d | K 11 = 420 K12 = 40 |
Further support for this explanation is provided by the determination of a solid-state structure (Fig. 6). Yellow crystals of [2]catenane 112+(Cl−)(PF6−) suitable for single crystal X-ray structural analysis were grown by slow evaporation of the 1:1 CDCl3:CD3OD titration solution of 112+(PF6−)2 and excess TBACl. The chloride anion sits in the interlocked cavity formed by the four amide and two para-pyridinium protons, while the hexafluorophosphate anion does not interact with the catenane. The local geometry of the catenane is less regular than that observed in the crystal structure of 102+(Cl−)(PF6−).11 While both of the pyridinium rings of 102+(Cl−)(PF6−) form face-to-face interactions with the hydroquinone groups of the other macrocycle, this is true of only one of the pyridinium rings in 112+(Cl−)(PF6−); the other forms a face-to-face contact with one hydroquinone ring, but is significantly non-parallel with the second (the angle between the best planes of the rings being 32.2°).
![The crystal structure of [2]catenane 112+(Cl−)(PF6−), where the chloride anion resides inside the cavity. Hydrogen atoms (except for those involved in hydrogen bonding to the chloride anion) have been omitted.](/image/article/2011/RA/c1ra00394a/c1ra00394a-f6.gif) | ||
| Fig. 6 The crystal structure of [2]catenane 112+(Cl−)(PF6−), where the chloride anion resides inside the cavity. Hydrogen atoms (except for those involved in hydrogen bonding to the chloride anion) have been omitted. | ||
The production of artificial receptors capable of binding anions in aqueous conditions is highly desirable considering, for example, the important biological roles anions possess.17 We have therefore begun considering the ability of our interlocked systems to achieve binding in aqueous solvent media, and very recently reported a dicationic rotaxane capable of binding chloride in solvent mixtures containing 35% D2O.18 Considering the very large association constant for chloride with catenane 112+(PF6−)2, titrations in an aqueous solvent mixture – CD3CN:D2O (70:30) were investigated (see Fig. 7 and Table 3). Upon the addition of chloride to catenane 112+(PF6−)2 downfield shifts of the pyridinium protons were observed consistent with the binding of the anion by the catenane, whereas only negligible movement (<0.02 ppm) in these resonances were observed upon the addition of dihydrogen phosphate and acetate, implying no binding of these anions. This last observation, combined with the calculated association constant for chloride of 230 M−1 in 30% water, is a testament to the affinity of the catenane for this anion. The [2]catenane 112+(PF6−)2 therefore exhibits remarkable selectivity for binding chloride over more basic singly charged anions in aqueous solvent systems.
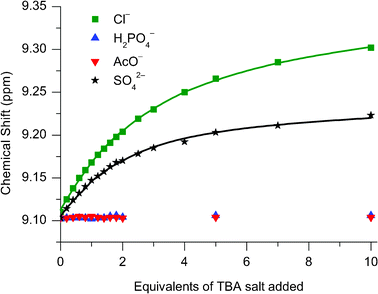 | ||
| Fig. 7
1H NMR titration data of 112+(PF6−)2 against TBA salts of Cl−, H2PO4− , AcO− and SO42− monitoring the para-pyridinium peak. Solvent: 70:30 CD3CN:D2O. T = 293 K. | ||
:30 CD3CN:D2Oa
A titration was also carried out with TBA2SO4 and 112+(PF6−)2 in CD3CN:D2O (70:30) (see Fig. 7 and Table 3). The calculated association constant was greater than that observed with chloride, however, considering the sulfate anion is doubly charged the magnitude difference is much smaller than might have been expected on purely electrostatic grounds. Further insight into the sulfate catenane association was provided by a X-ray structural analysis of a crystal of 112+(SO42−) grown from the diffusion of diisopropyl ether into a CH2Cl2/CH3OH solution of 112+(PF6−)2 and excess TBA2SO4. The solved structure (Fig. 8) shows the sulfate anion residing outside the catenane’s interlocked cavity, thus providing evidence that this polyatomic anion is unable to penetrate the interlocked cavity and associates peripherally instead. Hence sulfate is bound only marginally more strongly than the monoatomic chloride anion despite possessing double the charge. In this solid-state structure, the catenane pyridinium groups are arranged syn–anti, to allow for inter-ring hydrogen bonding (Fig. 8a). Meanwhile the sulfate counter-anion is found displaced from the interlocked organic fragment (Fig. 8b). Partial protonation of sulfate (to hydrogen sulfate) has occurred: probably from deprotonation of water molecules present in the solvent used to grow the crystals.
![The crystal structure of [2]catenane 112+(SO42−): (a) the [2]catenane structure, with the intramolecular amide–amide hydrogen bond depicted as dashed lines and (b) the location of the oxoanion counter-anions with respect to one of the symmetry identical macrocycles of the [2]catenane. Thermal ellipsoids are displayed at 50% probability.](/image/article/2011/RA/c1ra00394a/c1ra00394a-f8.gif) | ||
| Fig. 8 The crystal structure of [2]catenane 112+(SO42−): (a) the [2]catenane structure, with the intramolecular amide–amide hydrogen bond depicted as dashed lines and (b) the location of the oxoanion counter-anions with respect to one of the symmetry identical macrocycles of the [2]catenane. Thermal ellipsoids are displayed at 50% probability. | ||
Molecular dynamics simulations
In order to provide further insights into the anion recognition properties of the catenane 112+(PF6−)2 (Tables 2 and 3), molecular dynamics (MD) simulations were performed on the interlocked host’s association with Cl−, AcO− and H2PO4−.19Modelling in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1 CHCl3:CH3OH
:1 CHCl3:CH3OH
Using the X-ray structure of 112+(Cl−)(PF6−) as a starting geometry, the catenane was immersed in a cubic solvent box containing a 1:1 chloroform–methanol mixture, with structural data being collected during a 25 ns simulation. For the associations of the oxoanions AcO− and H2PO4− with the catenane, structures obtained from gas-phase quenched dynamic simulations were used. Representative co-conformations for the anion/catenane 112+ associations obtained from clustering the MD trajectories are presented in Fig. 9.
 | ||
| Fig. 9 Representative co-conformations of (a) 112+(Cl−), (b) 112+(AcO−), and (c) 112+(H2PO4−) in a 1:1 chloroform-methanol solution. Solvent molecules and the PF6− counter ion are omitted for clarity. Relevant hydrogen bonds are represented by dashed lines. The C–H hydrogen atoms (apart from N-methyl protons) have been omitted. | ||
The representative snapshot of catenane 112+(Cl−) in solution (Fig. 9a) shows the chloride anion residing in the tetrahedral binding cavity (defined by the four amide binding sites), establishing four N–H⋯Cl hydrogen bonds and presenting a remarkable similarity with the X-ray structure presented in Fig. 6. The N-methyl pyridinium rings are sandwiched between the hydroquinone rings of the other macrocycle establishing π–π interactions which is consistent with the experimental structural findings. The replacement of the monoatomic chloride anion by the polyatomic acetate (Fig. 9b) results in a severe distortion of the tetrahedral binding cavity in order to accommodate the carboxylate group of the anion. Both carboxylate oxygen atoms interact with amide binding sites, but only one is hydrogen bonded simultaneously to two N–H protons of a pyridinium cleft. The methyl head is outside the cavity and the face-to-face π–π stacking present in 112+(Cl−) is disrupted. Comparable structural outcomes are also observed in 112+(H2PO4−) with the anion establishing P–O(H)⋯H–N interactions with both macrocycles (Fig. 9c). However, because of its large stereoelectronic size, and in order to maximize the number of possible hydrogen bonds, the distortion is even larger than the one found for 112+(AcO−).
The stability of these binding arrangements in this solution mixture may be evaluated by monitoring the intermolecular distances between the centre of mass of the binding pocket defined by the four nitrogen atoms of the N–H binding sites (Crec) and Cl−, or the centre of mass (excluding the hydrogen atoms) of polyatomic anions AcO− and H2PO4− (Canion), over the duration of the simulation. The evolution of (Crec⋯Canion) distances during the course of MD simulations is plotted in Fig. 10 and the corresponding average distances are given in Table 4.
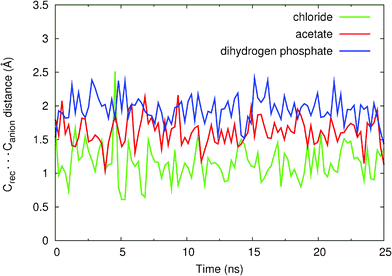 | ||
| Fig. 10 Variations in Crec⋯Canion intermolecular distances during the 25 ns of simulation for the binding associations of 11+ with Cl− (green), H2PO4− (blue) and AcO− (red) anions. The data was smoothed using a cubic spline interpolation. | ||
As can be seen in Fig. 10, the binding associations of 112+ with chloride, acetate and dihydrogen phosphate are stable during the 25 ns of simulation, the anions being kept hydrogen-bonded, having only short periods during which some of these bonds were interrupted.
Due to the nature of the chloroform/methanol solvent mixture and the differences between anion topologies, the calculation of absolute, or even relative, association constants is extremely difficult using standard approaches such as MM–PBSA (Molecular Mechanics–Poisson Boltzmann Surface Area) calculations or thermodynamic integration. However, we attempt here to give a qualitative description of the binding preferences of 112+ by analysing selected structural parameters taken from the simulations.
It is clear that in 112+(Cl−) the chloride anion is buried inside the catenane binding pocket as can be inferred by the short Crec⋯Canion average distance (1.13 Å) and Fig. 9a. Moreover, a hydrogen bond analysis performed on the entire MD trajectory data (Table S4, see ESI†) shows that the average percentage occupancies (the percentage of time in which the hydrogen bond is formed throughout the course of MD simulation) of the four N–H⋯Cl− bonds is greater than 90%, thus confirming the higher experimental association constant (K > 104 M−1) which corresponds to a larger experimental binding free energy (ΔG < −5.5 kcal mol−1).
The molecular recognition of oxoanions AcO− and H2PO4− by 112+, owing to their larger size, occurs mainly away from the centre of the binding pocket, with Crec⋯Canion average distances of 1.96 Å and 2.06 Å, respectively. The oxoanions possess specific geometric requirements to allow for simultaneous hydrogen bonding to all four amide groups, unlike the spherical chloride anion. The hydrogen bond analysis performed for 112+(AcO−) and reported in Table S5 (see ESI†) indicates that one acetate oxygen is participating in hydrogen bonding with three N–H binding groups (% occupancy > 84%). The remaining carboxylate oxygen is engaged only in one hydrogen bond (∼80% occupancy). The same type of analysis performed for 112+(H2PO4−) presented in Table S6 (see ESI†) indicates very similar results: one oxygen atom (–O) is involved in three hydrogen bonds with the N–H groups (% occupancy > 63%) while the other –O participates in only one strong hydrogen bond (70% occupancy). The –OH groups of H2PO4− can also bind the N–H groups reaching a maximum of occupancy of 33%. These values obtained for 112+(AcO−) and 112+(H2PO4−) are lower than the ones found for 112+(Cl−), thus clearly demonstrating the higher affinity of catenane 112+ towards chloride. The relative binding preferences between AcO− and H2PO4− are not as easily explained. Apparently, the theoretical findings seem to indicate a preference towards AcO−, which is not in agreement with the experimental data presented in Table 2. However, if one takes into account the more spherical topology of H2PO4−, it can swap more easily between N–H binding sites and this is indeed reflected if one sums the occupancy percentages of donor atoms. In other words the N–H bonds are more occupied in H2PO4− than in AcO−. It should be pointed out that the different association constants (K(AcO−) = 160 M−1 and K(H2PO4−) = 1240 M−1) correspond to ΔG(AcO−) = −3.0 kcal mol−1 and ΔG(H2PO4−) = −4.2 kcal mol−1. The difference in free energies, δΔG = 1.2 kcal mol−1, may be too small for the accuracy of the anion parameters, which were not specifically developed for this type of solvent mixtures.
Modelling in 70:30 CH3CN:H2O
As shown in Table 3, catenane 112+(PF6−)2 shows a remarkable selectivity for chloride in the solvent system containing 30% water, but with a notably lower association constant. This inspired us to perform MD simulations on 112+(Cl−) in a CH3CN:H2O (70:30) mixture using the same procedure as followed for the chloroform–methanol mixture. Again, in order to check the stability of the 112+(Cl−) binding arrangement, the distance between the anion (Canion) and the centre of mass of the four nitrogen atoms of the N–H binding sites (Crec) was monitored during the 25 ns of simulation time. The Crec⋯Canion distances, plotted in Fig. 11, indicate that 112+(Cl−), is stable for 8.5 ns with an average Crec⋯Canion distance of 1.21 ± 0.29 Å (for n = 8500). After this time, the Crec⋯Canion distances start to increase and the chloride leaves the binding pocket of 112+. The water shell around the chloride, defined as the number of water molecules closer than 3.4 Å of the anion, is also plotted in Fig. 11. It can be seen that in this very competitive medium, the catenane is able to “protect” the anion from the water only for ∼8.5 ns and subsequently, the number of water molecules around chloride increase dramatically. The stability of the N–H⋯Cl− hydrogen bonds (% occupancy) was also measured in this system and the results are reported in Table S4 (see ESI†). Compared with the CHCl3:CH3OH simulation, in CH3CN:H2O the N–H⋯Cl− hydrogen bond occupancy has decreased dramatically to ∼30%. These simulations indicate that, although catenane 112+ is indeed able to bind chloride in this highly competitive medium, the strength of this binding is much lower than the one found in CHCl3:CH3OH which is in agreement with the experimental findings (K > 104 M−1vs. K = 230 M−1). Hence, these MD simulations are at least qualitatively consistent with the experimental data.
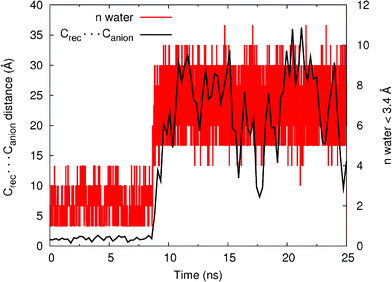 | ||
| Fig. 11 Variations in Crec⋯Canionintermolecular distances for the 25 ns of simulation for the binding associations of 11+ with Cl− (black) plotted against the number of water molecules closer than 3.4 Å to the chloride anion. The distance data was smoothed using a cubic spline interpolation. | ||
Conclusions
We have shown it is possible to synthesize anion templated [2]catenanes by the double clipping of two carefully designed acyclic pyridinium components around a chloride template, in very high yields. The interlocked nature of the systems was proven by a combination of 1H NMR spectroscopy, electrospray MS and X-ray crystallography. The cavities of the [2]catenanes were found to be highly selective for chloride over monocharged oxoanions, in sharp contrast to their precursors, with the ability to selectively bind chloride in a competitive aqueous solvent system containing 30% D2O. Importantly under these aqueous solvent conditions, no binding of the more basic monocharged oxoanions acetate and dihydrogen phosphate was observed. Molecular dynamic simulations revealed this may be due to the binding of the larger polyatomic anions leading to distortions of the catenane cavities which result in weakening of the π–π stacking between the pyridinium and hydroquinone rings.Acknowledgements
N. H. E. wishes to thank the EPSRC for a DTA studentship. M. D. L. wishes to thank the EPSRC and GE Healthcare for a CASE-supported studentship. C. J. S. also wishes to thank the EPSRC for a CASE sponsored studentship (in conjunction with Johnson–Matthey) and for post-doctoral funding (PhD Plus). K.-Y. N. wishes to thank the Clarendon Fund and the Overseas Research Student Awards Scheme for a scholarship. We are grateful to Diamond Light Source for the award of beamtime on I19 (MT1858) and to the beamline scientists for help and support. S. M. S. acknowledges FCT for a PhD grant (SFRH/BD/29596/2006). P.J.C. thanks FCT for the postdoctoral grant SFRH/BPD/27082/2006. V.F. acknowledges the financial support from FCT under the project PTDC/QUI/68582/2006 with co-participation of the European Community funds FEDER, QREN and COMPETE.References
-
(a)
V. Balzani, M. Venturi and A. Credi, Molecular Devices and Machines: A Journey into the Nanoworld, Wiley-VCH, Weinheim, 2003 CrossRef
; (b) E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72–191 CrossRef CAS
-
Molecular Catenanes, Rotaxanes and Knots: A Journey Through the World of Molecular Topology (ed.: J.-P. Sauvage and C. Dietrich-Buchecker), Wiley-VCH, Weinhelm, 1999 Search PubMed
-
(a) E. Wasserman, J. Am. Chem. Soc., 1960, 82, 4433–4434 CrossRef CAS
-
(a) C. O. Dietrich-Buchecker and J.-P. Sauvage, Chem. Rev., 1987, 87, 795–810 CrossRef CAS
-
(a) C. A. Hunter, J. Am. Chem. Soc., 1992, 114, 5303–5311 CrossRef CAS
-
(a)
J. L. Sessler, P. A. Gale and W-S. Cho, Anion Receptor Chemistry, RSC, Cambridge, 2006 Search PubMed
-
(a) J. A. Wisner, P. D. Beer and M. G. B. Drew, Angew. Chem., Int. Ed., 2001, 40, 3606–3609 CrossRef CAS
-
(a) J. A. Wisner, P. D. Beer, M. G. B. Drew and M. R. Sambrook, J. Am. Chem. Soc., 2002, 124, 12469–12476 CrossRef CAS
-
(a) M. R. Sambrook, P. D. Beer, J. A. Wisner, R. L. Paul and A. R. Cowley, J. Am. Chem. Soc., 2004, 126, 15364–15365 CrossRef CAS
-
(a) For examples of other research groups utilizing anion templation in the construction of interlocked structures, see: G. M. Hubner, J. Glaser, C. Seel and F. Vögtle, Angew. Chem., Int. Ed., 1999, 38, 383–386 CrossRef CAS
- The synthesis, characterization and anion binding studies of catenane 102+(X−)(Y−) have been previously reported in a preliminary communication: K.-Y. Ng, A. R. Cowley and P. D. Beer, Chem. Commun., 2006, 3676–3678 RSC
- A preliminary 1H NMR titration study in which 1+PF6− was titrated with TBACl in CDCl3 provides evidence for the formation of these two competing (2:1 and 1:1) binding modes. While pyridinium and amide protons move downfield throughout the titration (as expected upon anion binding), the maximum upfield shift and splitting of the hydroquinone protons (indicative of π–π stacking) occurred at 0.5 eq. of TBACl, with addition of further TBACl reducing the magnitude of peak separation, with the peaks also moving back downfield. This may be rationalized by the competing nature of the 2:1 and 1:1 binding modes, the former is more favourable when chloride concentration is up to half that of 1+PF6−, while any increase in chloride concentration thereafter favours the 1:1 binding mode, which reduces π–π stacking, and hence the splitting of the hydroquinone protons. For selected spectra from this titration, see ESI†.
- The assignment of this second catenane product is based on the expectation that the catenane will be formed with one equivalent of chloride bound in the cavity. Indeed, titrating TBACl into an NMR sample of this material into the very competitive 70:30 CD3CN:D2O solvent system revealed that while binding of this additional chloride was occurring, it was weaker than that of 112+(PF6−)2. While the isolated material definitively contains hexafluorophosphate (by the appearance of a doublet at 19F NMR spectrum), elemental analysis proved inconclusive, with satisfactory results only being obtained in the case of the dihexafluorophosphate salt 112+(PF6−)2.
-
(a) T. M. Trnka and R. H. Grubbs, Acc. Chem. Res., 2001, 34, 18–29 CrossRef CAS
- M. J. Hynes, J. Chem. Soc., Dalton Trans., 1993, 311–312 RSC
- The increased binding strength of an ionic guest by a catenane compared to its non-interlocked components was first demonstrated by the group of Jean-Pierre Sauvage, see: F. Arnaudeu, E. Marques, M. J. Schwingeill, C. Dietrich-Buchecker and J.-P. Sauvage, New J. Chem., 1988, 12, 15–20 Search PubMed
- S. Kubik, Chem. Soc. Rev., 2010, 39, 3648–3663 RSC
- L. M. Hancock, L. C. Gilday, S. Carvalho, P. J. Costa, V. Félix, C. J. Serpell, N. L. Kilah and P. D. Beer, Chem.–Eur. J., 2010, 16, 13082–13094 CrossRef CAS
- For full details of the computational methods used, along with some analogous modelling data of [2]catenane 102+(PF6−)2 see ESI.
Footnote |
| † Electronic Supplementary Information (ESI) available: Synthetic procedures and spectral characterization of novel compounds; crystallographic information; 1H NMR titration protocols; details of computational modelling, with additional data and discussion. CCDC reference numbers 832250- 832252. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1ra00394a/ |
| This journal is © The Royal Society of Chemistry 2011 |