Electrochemistry of graphene: not such a beneficial electrode material?†
Dale A. C.
Brownson
,
Lindsey J.
Munro
,
Dimitrios K.
Kampouris
and
Craig E.
Banks
*
Faculty of Science and Engineering, School of Science and the Environment, Division of Chemistry and Environmental Science, Manchester Metropolitan University, Chester Street, Manchester M1 5GD, Lancs, UK. E-mail: c.banks@mmu.ac.uk; Fax: ++(0)1612476831; Tel: ++(0)1612471196 Web: www.craigbanksresearch.com
First published on 8th September 2011
Abstract
We critically evaluate the reported electro-catalysis of graphene using inner-sphere and outer-sphere electrochemical redox probes, namely potassium ferrocyanide (II) and hexaammine-ruthenium(III) chloride, in addition to L-ascorbic acid and β-nicotinamide adenine dinucleotide. Well characterised commercially available graphene is utilised which has not been chemically treated, is free from surfactants, and as a result of its fabrication has an extremely low oxygen content allowing the electronic properties to be properly de-convoluted. Surprisingly we observe that graphene exhibits slow electron transfer towards the electrochemical probes studied, effectively blocking underlying electron transfer of the supporting electrode substrate likely due to its large basal and low edge plane content. Such observations, never reported before, suggest that graphene may not be such a beneficial electrode material as widely reported in the literature. Density Functional Theory is conducted on symmetric graphene flakes of varying sizes indicating that the HOMO and LUMO energies are concentrated around the edge of the graphene sheet, at the edge plane sites, rather than the central basal plane region, consistent with experimental observations. We define differentiating coverage-based working regions for the electrochemical utilisation of graphene: ‘Zone I’, where graphene additions do not result in complete coverage of the underlying electrode and thus increasing basal contribution from graphene modification leads to increasingly reduced electron transfer and electrochemical activity; ‘Zone II’, once complete single-layer coverage is achieved, layered graphenevizgraphite materialises with increased edge plane content and thus an increase in heterogeneous electron transfer is observed with increased layering. We offer insight into the electrochemical properties of these carbon materials, invaluable where electrode design for electrochemical sensing applications is sought.
Introduction
Graphene, a single atomic layer thick two-dimensional carbon nanomaterial, has been reported to possess spectacular physical, chemical, and electrical properties,1–4 and has consequently received enormous interest from a plethora of scientific disciplines into the exploration and exploitation of its unique properties.4 One area of particular interest where graphene has impacted significantly is electrochemistry, where it has been widely used as an electrode material within a variety of sensing and energy related devices, claiming superior electrochemical performances when compared to traditional noble metals and various fullerene based electrode materials, such as graphite and carbon nanotubes.2,3,5Graphene's popularity has resulted in its application in the development of significantly enhanced energy storage and generation utilities, for example, in lithium based rechargeable batteries,6 super-capacitors,7 and in a variety of interesting fuel cell devices.8,9Graphene has also been reported to exhibit enhanced performance towards the sensing of acetaminophen,10dopamine,11glucose,12hydrazine,13 and nitric oxide14 to name just a few.
While there are many optimistic reports of utilising graphene, reports have emerged demonstrating that graphene might not provide a significant advantage over existing electrode materials.15–18 Pumera et al.15 has elegantly shown that single-, few-, and multi-layer graphene does not exhibit a significant advantage over graphite micro-particles in terms of sensitivity, linearity, and repeatability towards the electro-analytical detection of uric acid. Recently it has been shown that surfactants routinely used in the production of commercially available graphene both detrimentally and beneficially interfere towards different analytes and are a major contribution to the observed electrochemical response, potentially leading to false claims of the electro-catalysis of graphene,16,17 preventing the true heterogeneous electron transfer of graphene to be observed and its electronic properties to be de-convoluted.
Notably, it has recently been demonstrated that the origin of electron transfer in graphene is from its peripheral edge, as opposed to its side,19 where the former acts electrochemically akin to that of edge plane- and the latter to that of basal plane- like- sites/defects of highly ordered pyrolytic graphite (HOPG). Note that in this model, the electron transfer kinetics of the edge are overwhelmingly dominant over that of the basal. However, recent evidence for graphitic surfaces has shown that in certain cases the basal plane has a measurable activity.20 Thus in certain limited cases it might be likely our model breaks down and the side of graphene, which is basal plane like, might exhibit measurable activity in these restricted cases.
In this paper we explore for the first time the electronic structure of graphene which is commercially available and well characterised—produced via a substrate-free gas-phase synthesis method.21–23 The graphene is completely free from surfactants and is not purposely oxidised or treated and consequently has ∼4.96% oxygen content, indicating near-true graphene. Fig. 1 shows a high-resolution TEM image in addition to an atomic-resolution image that clearly reveal a highly ordered synthesised single-layer graphene sheet: a conceptual image of graphene's physical structure indicating it's basal and edge plane electron transfer sites is also shown. The effect of coverage when utilising graphene as an electrode material is explored towards both inner-sphere and outer-sphere electrochemical redox probes, namely potassium ferrocyanide (II) and hexaammine-ruthenium(III) chloride, in addition to L-ascorbic acid and β-nicotinamide adenine dinucleotide, with the responses compared and contrasted towards graphite alternatives.
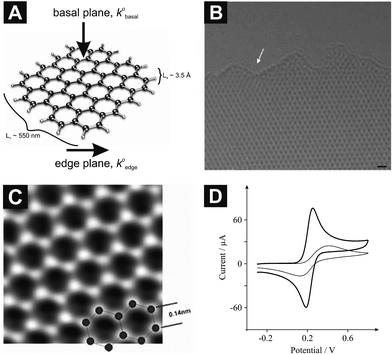 | ||
| Fig. 1 (A) Schematic representation of graphene indicating its edge and basal sites, where the heterogeneous electron transfer rate of the former (k0edge) is anomalously faster over that of the latter (k0basal), which constitutes the largest contribution to its surface area. Also shown are the relative sizes as indicated by the intraplanar microcrystallite size, La, and interplanar microcrystallite size, Lc. (B) A high-resolution TEM image, where the white arrow indicates the edge of the graphene sheet; scale bar is 4 Å. (C) An atomic-resolution image (TEAM 0.5) of a clean and structurally perfect synthesized graphene sheet. Individual carbon atoms appear white in the image. (D) Cyclic voltammetric profiles recorded for 1 mM potassium ferrocyanide (II) in 1 M KCl using an EPPG electrode (thick line), and a BPPG electrode (thin line): Scan rate: 100 mVs−1 (vs.SCE). B and C are reproduced with permission from Ref. 22. | ||
We demonstrate for the first time that there are two essential coverage regions for graphene, where at ‘Zone I’ low global coverage of graphene, Θgraphene, immobilised onto an electrode substrate exhibits slow heterogeneous electron transfer owing to a large basal plane content. Conversely in ‘Zone II’, equating to high coverage of graphene, fast heterogeneous electron transfer is observed as deviation from purely single layered graphene to few- and multi- layered graphene leads to increased edge plane content. This work shows that in the cases explored here, graphene exhibits slow electrode kinetics due to its low edge-to-basal plane ratio and consequently graphite appears to be a superior electrode material.
Experimental section
All chemicals were of analytical grade (or higher) and were used as received from Sigma-Aldrich without any further purification. All solutions were prepared with deionised water of resistivity not less than 18.2 MΩ cm and were vigorously degassed with high purity, oxygen free nitrogen prior to electrochemical measurements. Voltammetric measurements were carried out using a μ-Autolab III (ECO-Chemie, The Netherlands) potentiostat. All measurements were conducted using a three electrode system. The edge plane pyrolytic graphite (EPPG) working electrode (Le Carbone, Ltd. Sussex, U.K) was machined into a 4.9 mm diameter, with the disc face parallel with the edge plane as required from a slab of HOPG (highest grade available: SPI-1, equivalent to Union Carbide's ZYA grade, with a lateral grain size, La of 1–10 μm and 0.4 ± 0.10 mosaic spread); alternatively, the basal plane pyrolytic graphite (BPPG) working electrode (4.9 mm diameter, Le Carbone, Ltd. Sussex, U.K.) was machined as above however with the disc face parallel to the basal plane as required. A platinum wire and a saturated calomel electrode (SCE) were used as counter and reference electrodes respectively.The graphene was commercially obtained from ‘Graphene Supermarket’ (Reading, MA, USA), and are known as ‘Pristine Graphene Monolayer Flakes’, comprising entirely of pristine graphene platelets dispersed in ethanol (solutions consisted of either 10 μg per 10 mL or 0.5 mg per 10 mL) that have not been oxidised, reduced, or chemically modified in anyway and are free from surfactants.24 The graphene was synthesised via the substrate-free gas-phase method, as previously reported,21–23 and are sonicated in ethanol to form a homogeneous suspension before being dispatched.23 Further details concerning the fabrication process are provided in the ESI.† The graphene has an average flake thickness of 0.35 nm (1 monolayer) with an average particle (lateral) size of 550 nm (150–3000 nm).24XPS chemical analyses were performed with a VG-Microtech Multilab electron spectrometer and revealed the material to comprise of 95.04% atomic carbon and 4.96% atomic oxygen; the low O/C ratio indicates near true graphene.
Aliquots of the graphene were carefully pipetted onto the electrode surface using a micro-pipette and allowed to dry at room temperature under nitrogen flow in order to eliminate oxidation of the graphene by the presence of atmospheric oxygen, following which the electrode could either be further modified, or was ready to use. All comparison and control measurements using graphite or ethanol were conducted within the exact manner as with graphene: two separate solutions of graphite dispersed in ethanol were utilised, dilute graphite (1 μg per 2 μL), and concentrated graphite (100 μg per 1 μL). Note, control experiments were performed in terms of ethanol modified electrodes for the purpose of de-convoluting the origin of the electro-activity and ensuring that electrochemical responses observed were not a result of the solvents utilised; such control experiments revealed that ethanol has no effect upon electro-activity.
Spin-polarised density-functional theory (DFT) calculations were performed using the ORCA program.25 The geometries of all of the structures were optimised using the B3LYP hybrid exchange–correlation functional and a 6-31G** basis set,26–28 where the optimisation was terminated when the magnitude of the force on each atom was less than 0.015 eV Å−1.
Results and discussion
Results
We first characterise the electron transfer properties of edge and basal plane electrodes, constructed from HOPG, evaluated with the inner-sphere electron transfer redox probe,29 1 mM potassium ferrocyanide (II) in 1 M KCl. Fig. 1D depicts characteristic voltammetric profiles obtained at EPPG and BPPG electrodes where the former exhibits a peak-to-peak potential separation, ΔEp, of 60 mV (at 100 mVs−1) while the latter exhibits a ΔEp of 242 mV (at 100 mVs−1). Both responses are in excellent agreement with previous studies19,29–31 since the edge plane electrode has a high global coverage of edge plane sites which exhibit anomalously fast electron transfer rates over that of basal plane sites.32 Conversely, the BPPG electrode due to its structure has a low global coverage of edge plane sites and hence a poor voltammetric activity is exhibited.32 Attention was next turned to exploring the cyclic voltammetric performance of an EPPG electrode following modification with 40 ng of graphene (see Experimental section). As depicted in Fig. 2A the introduction of graphene results in a change to the peak-to-peak separation from 60 to 192 mV (at 100 mVs−1), which is similar to that observed for the unmodified BPPG electrode (242 mV; 100 mVs−1). Note that this approach, where an electrode material exhibiting fast electron transfer rates is modified with graphene, is commonly utilised in the literature.10,33 Next we consider the effects of graphene coverage on the electrochemical response. Fig. 2A depicts the increasing mass deposition of graphene onto an underlying EPPG electrode surface where it is clear that with increasing graphene coverage, a decrease in the voltammetric peak height is evident as well as a significant decrease in the electrochemical reversibility of the redox probe, as evident by the increasing ΔEp. Note that this response is distinctly different to all current literature regarding graphene.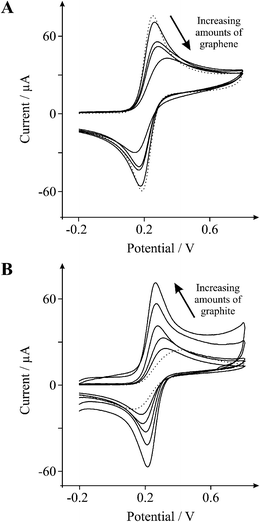 | ||
| Fig. 2 Cyclic voltammetric profiles recorded utilising 1 mM potassium ferrocyanide (II) in 1 M KCl. A: Cyclic voltammetric profiles obtained using an EPPG electrode (dotted line) with the addition of increasing amounts of 10, 20, 30, and 40 ng graphene (solid lines). B: Cyclic voltammetric profiles obtained using a BPPG electrode (dotted line) with the addition of increasing amounts of 2, 4, 50, 100, and 200 μg graphite (solid lines). Scan rate: 100 mVs−1 (vs.SCE). | ||
For comparative purposes we next turn to exploring the cyclic voltammetric performance of a BPPG electrode following modification with 50 μg of graphite (as above). As evident in Fig. 2B the graphite modified electrode exhibits a ΔEp of 66 mV (at 100 mVs−1), which is similar to that observed at an EPPG electrode (60 mV; 100 mVs−1) and significantly smaller than that observed at both the graphene modified EPPG and the underlying BPPG electrodes (192, and 242 mV respectively; 100 mVs−1). Note that this response is as commonly observed in the literature. Through comparison of the peak-to-peak separations it is clear that modification with graphite leads to an increased proportion of edge plane sites (electron transfer sites) and thus results in an enhanced electrochemical response with increased reaction kinetics, reversibility and electro-catalytic activity over that of the underlying BPPG electrode and a graphene modified EPPG electrode (see earlier).32,34–37
We next characterise the electronic properties of an EPPG electrode using the outer-sphere electron transfer redox probe,29 1 mM hexaammine-ruthenium(III) chloride in 1 M KCl. Fig. 3A depicts a typical cyclic voltammetric profile obtained at an EPPG electrode where a ΔEp of 60 mV (at 100 mVs−1) is observed, which is in excellent agreement with previous literature.38 Also shown in Fig. 3A is the cyclic voltammetric response obtained as a result of modification of the underlying EPPG electrode with 200 ng graphene, where an increase in the ΔEp occurs from 60 (EPPG) to 115 mV (after modification with 200 ng graphene) at 100 mVs−1 which is characteristically similar to that reported for a BPPG electrode.38 The effect of increasing graphene's mass deposition on the underlying EPPG electrode is also depicted in Fig. 3A where with an increased coverage of graphene the electron transfer kinetics (viz reversibility of the redox probe) are significantly reduced, thus after modifying an EPPG electrode with 400 ng of graphene a ΔEp of 141 mV is observed. Fig. 3B depicts a plot of ‘ΔEp’ versus ‘mass of graphene deposited’ which reveals a positive correlation where an increase in the mass of graphene results in an increase in the ΔEp; again such a response has never been reported before in the literature regarding graphene.
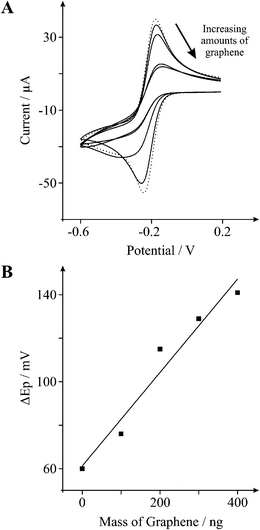 | ||
| Fig. 3 A: Cyclic voltammetric profiles recorded utilising 1 mM hexaammine-ruthenium(III) chloride in 1 M KCl, obtained using an EPPG electrode (dotted line) with the addition of increasing amounts of 100, 200, 300, and 400 ng graphene (solid lines). Scan rate: 100 mVs−1 (vs.SCE). B: Relationship between the mass of graphene deposited upon the electrode surface and the resultant peak-to-peak separation. | ||
Now we turn to exploring the electrochemical properties of graphene within bio-sensing applications and first consider the effect of various electrode modifications towards the sensing of 1 mM β-nicotinamide adenine dinucleotide (NADH) in pH 7 phosphate buffer solution (PBS) using both EPPG and BPPG electrodes. Fig. 4 shows a typical voltammetric response towards NADH utilising both EPPG and BPPG electrodes where the oxidation peak potentials occur at ∼490 and ∼660 mV respectively and are in excellent agreement with a previous literature report.36 In the former, a voltammetric signature is observed at a lower (positive) over-potential than that observed for the latter, which is indicative of a greater reactivity on the EPPG over that of the BPPG electrode due to a larger global coverage of edge plane sites,32 which represents increased heterogeneous electron transfer kinetics at the EPPG electrode.
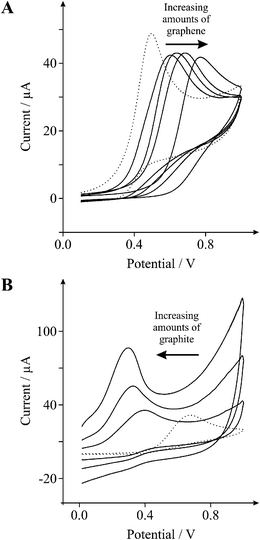 | ||
| Fig. 4 Cyclic voltammetric profiles recorded utilising 1 mM NADH in PBS (pH 7). A: Cyclic voltammetric profiles obtained using an EPPG electrode (dotted line) with the addition of increasing amounts of 2.5, 5.0, 10.0, and 40.0 ng graphene (solid lines). B: Cyclic voltammetric profiles obtained using a BPPG electrode (dotted line) with the addition of increasing amounts of 50, 100, and 200 μg graphite (solid lines). Scan rate: 100 mVs−1 (vs.SCE). | ||
Attention was next turned to exploring the cyclic voltammetric performance of an EPPG electrode following modification with 10 ng of graphene, again, common practice in the current graphene literature. As depicted in Fig. 4A the introduction of graphene results in a positive shift in the oxidative peak potential from ∼490 (EPPG) to ∼665 mV (10 ng graphene), which is characteristic of an unmodified BPPG electrode (∼660 mV), as observed above. When considering the effect of graphene coverage on the voltammetric response (shown in Fig. 4A) it is evident that increasing the mass of graphene deposited results in a clear shift in the voltammetric over-potential (oxidation peak) of NADH towards more electropositive regions, where at 40 ng of graphene the oxidation peak occurs at ∼772 mV, indicating slow heterogeneous electron transfer at graphene.
We now turn to exploring the effect of graphite upon a BPPG electrode, where as evident in Fig. 4B the addition of 50 μg of graphite onto the BPPG electrode surface results in a negative shift in the oxidative peak potential from ∼660 (BPPG) to ∼380 mV (50 μg graphite), which is characteristically similar to that of an unmodified EPPG electrode (∼490 mV). With the addition of increasing quantities of graphite onto the BPPG electrode a further shift of the NADH oxidation peak is apparent, displaying continuous movement towards less electropositive over-potentials, where after the addition of 200 μg graphite onto the underlying BPPG electrode surface an oxidation peak occurs at ∼292 mV, indicating fast heterogeneous electron transfer at graphite; such a response is routinely reported in the literature.
To further explore the effects of graphene upon various biologically relevant compounds we next explore the electronic properties of graphene using 1 mM L-ascorbic acid in PBS (pH 7). Fig. 5A depicts the standard cyclic voltammetric response observed at an EPPG electrode where an oxidation peak is evident at ∼227 mV. Upon modification of the EPPG electrode with 20 ng of graphene there is a positive shift in the oxidative peak potential (over-potential) from ∼227 (EPPG) to ∼543 mV (20 ng graphene), which is characteristic of a BPPG electrode.39Fig. 5B shows the effect of increasing graphene coverage on the position of the oxidation peak for L-ascorbic acid, where a positive correlation indicates that an increased mass of graphene deposited upon the underlying electrode surface results in a further shift in the oxidative over-potential towards electropositive regions, where after the addition of 10, 20, and 40 ng of graphene the oxidation potential shifts to ∼363, ∼543, and ∼794 mV respectively; for comparative purposes note that the oxidation potential was ∼386 mV for an unmodified BPPG electrode.
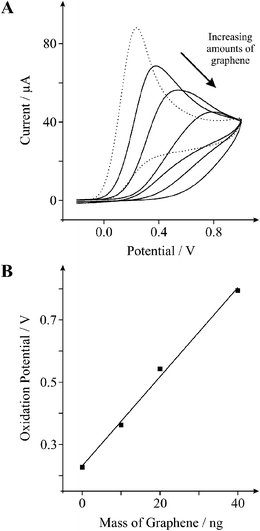 | ||
| Fig. 5 A: Cyclic voltammetric profiles recorded utilising 1 mM L-ascorbic acid in PBS (pH 7) obtained using an EPPG electrode (dotted line) with the addition of increasing amounts of 10, 20, and 40 ng graphene (solid lines). Scan rate: 100 mVs−1 (vs.SCE). B: Relationship between the mass of graphene deposited upon the electrode surface and the resultant oxidation peak potential. | ||
Discussion
Returning to the response observed in Fig. 3 where the addition of graphene results in greater peak-to-peak separations, if we assume the response is akin to that of a partially blocked electrode as identified by Amatore and co-workers,40 the observed heterogeneous electron transfer rate kinetics, k0obs, are related to the electron transfer rate of the unmodified electrode, k0UE, and the fractional coverage of graphene, θ, by:| k0obs = k0UE(1 − θ) | (1) |
| θR = 1 − e−θ | (2) |
Comparison of the standard heterogeneous electron transfer rates indicate that the addition of graphene onto the EPPG electrode results in the reduction of the observed electron transfer kinetics. It is well documented that the voltammetric response of graphitic electrodes depends on the proportion of edge plane sites where a low and high proportion result in slow and fast electron transfer respectively,34,45 which has also recently been shown to be the case for graphene.19 Consequently, where graphene was utilised earlier the deterioration in the electrochemical responses observed are likely due to the reduced proportion of available edge plane sites and an increased basal plane contribution from the addition of graphene (which is basal plane abundant by nature). Note that the range of electro-active species studied here allow the contribution from the oxygenated species residing on the edge plane of the graphene to be neglected since the observed trend is the same for all compounds studied which range from simple outer-sphere electron transfer probes to surface sensitive inner-sphere species.45
To further understand and highlight the contribution of edge and basal plane sites which are an inherent property of graphene and graphite alike, let us now consider the surface area ratio of basal plane, Sbasal, to edge plane (perimetrical boundary carbon atoms), Sedge, for the case of increasing layers of graphene. The mean surface area of the basal contribution for a single piece of graphene may be deduced using Sbasal = πr2, where for the graphene utilised in this study the radius, r, is ∼275 nm. The surface area of the edge plane for a single piece of graphene is deduced using Sedge = 2πrh, where the Sedge is considered to be a round strip or band consisting of the boundary of carbon atoms of the graphene layer, the same as the length of the perimeter of a circular disk, and the width is the height of this stripe (h)—thus for a single layer of graphene the height of the strip is equal to the thickness of the monolayer sheet, which is 3.5 Å, and each layer of graphene added contributes to an increase in the Sedge by 604.76 nm2.18,34,45 Note, the assumption that graphene sheets are circular is readily for exemplification and will likely not be exactly the case in true graphene. Fig. 6 compares the ratio of Sbasal/Sedge on increasing the number of graphene layers where it is evident that as more layers are introduced, the amount of edge, viz edge plane sites is increased. Additionally, this is related to the theoretical current that would be expected as the amount of edge plane is increased, assuming the Sedge to be a ‘band type’ electrode, where the edge of the graphene is effectively the surface area of the electrode and as a consequence of increasing the amount of edge plane an increase in the current is expected.18
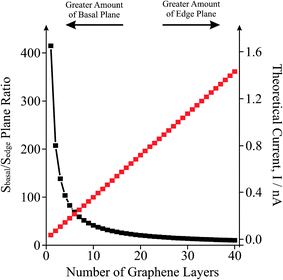 | ||
| Fig. 6 The effect of increasing the number of graphene layers upon the ratio of basal/edge plane sites for a single piece of graphene (black squares) and the effect on the theoretical observed current (red squares). Note that this is for a single piece of graphene. | ||
Thus as we move from single-layer graphene to multi-layer graphene, which is approaching that of graphite, the observed voltammetric responses will ‘theoretically’ surpass that of graphene. Note that this representation considers a single piece of graphene for simplicity, but yet it is insightful. However in a real experimental case the graphene will likely be immobilised onto an underlying electrode surface and assuming that graphene exhibits fast electron transfer kinetics over that of the underlying electrode, the response of the electrode would be akin to that of an electrode array and the current response would need to multiplied, as best can be estimated, for the total number of graphene pieces comprising the electrode assuming that there is no physical interaction of the graphene domains and that they are sufficiently separated from their nearest neighbour, meeting the voltammetric requirements of diffusional independence.46,47 Note that in reality there will be some degree of overlap and the diffusion domain approach pioneered by Compton46,48 would ideally be suited and could be adapted to include the variance in the graphene from true graphene, viz single layer, to few layer graphene.
It is evident that modification of an electrode surface with nanogram quantities of graphene leads to incomplete ‘single layer’ coverage of the underlying electrode surface where the ‘uncovered’ underlying electrode material remains electrochemically active (vizEPPG electrode) or relatively inactive (vizBPPG electrode) depending on the material utilised. In our case the high global coverage of edge plane sites on the underlying EPPG electrode exhibit faster heterogeneous electron transfer kinetics than that of the basal plane surface area of graphene (which is electrochemically inert in contrast to edge plane sites), thus with increased coverage of graphene up to one ‘complete single layer’ the underlying material is increasingly inhibited and consequently slower heterogeneous electron transfer resides, induced from the basal plane sites of graphene now dominating the electrochemical reactivity and ‘blocking’ the underlying edge plane sites of the host material, as evident with the continual increase in the observed ΔEp values in the case of potassium ferrocyanide (II) for example (Fig. 2A).
Interestingly, when larger quantities of graphene are utilised (micrograms opposed to nanograms), as evident in Fig. 7A, it is clear that the electrochemical reversibility and respective heterogeneous electron transfer rate begins to increase with increased mass deposition of graphene (ΔEp of 155, 128, and 123 mV for the addition of 0.5, 1.0, and 2.0 μg of graphene respectively (at 100 mVs−1)). This is additionally confirmed utilising the biologically relevant compound, 1 mM L-ascorbic acid in PBS (pH 7) where as depicted in Fig. 7B the modification of 20 and 100 ng of graphene upon an EPPG electrode leads to the continual reduction in electrode rate kinetics and consequently there is an evident shift in the over-potential to higher electropositive regions, with the oxidation peak potential for an unmodified EPPG electrode and EPPG electrodes modified with 20 and 100 ng of graphene residing at ∼195, ∼441, and ∼776 mV respectively. However, when larger quantities of graphene are added (1.0 and 1.5 μg) the over-potential is significantly reduced, resulting in a shift towards lower electropositive regions, and thus the peak potential is observed at ∼422 and ∼383 mV respectively. This trend is also exemplified in Fig. 7C where with large microgram masses of graphene, the oxidation potential shifts beneficially to less positive potentials.
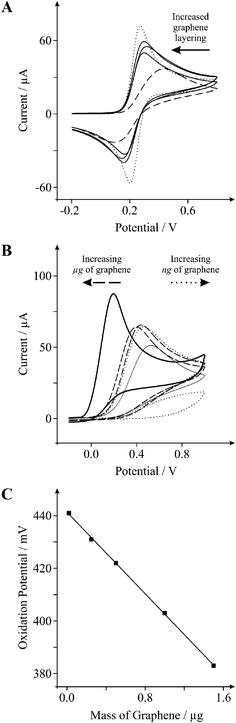 | ||
| Fig. 7 A: Cyclic voltammetric profiles recorded utilising 1 mM potassium ferrocyanide (II) in 1 M KCl, obtained using an EPPG electrode (dotted line) with the addition of increasing amounts of 0.5, 1.0, and 2.0 μg graphene (solid lines), and using a BPPG electrode (dashed line). B: Cyclic voltammetric profiles recorded for 1 mM L-ascorbic acid in PBS (pH 7) utilising a BPPG electrode (thin solid line), and an EPPG electrode (thick solid line) with the addition of increasing amounts of; 20 and 100 ng graphene (dotted lines), and 1.0 and 1.5 μg graphene (dashed lines). C: Relationship between; the mass of graphene deposited upon the electrode surface and the resultant oxidation peak potential of 1 mM L-ascorbic acid in PBS (pH 7). Scan rates: 100 mVs−1 (vs.SCE). | ||
From the above observations we are able to define differentiating coverage-based working regions for the electrochemical utilisation of graphene: ‘Zone I’, where graphene additions do not result in complete coverage of the underlying electrode surface and thus increasing basal contribution from the graphene modification leads to increasingly reduced electron transfer kinetics and electrochemical reactivity; ‘Zone II’, once complete single-layer coverage is achieved, layered graphenevizgraphite materialises with increased global edge plane content and thus an increase in heterogeneous electron transfer is observed with increased layering, evident via an improvement in the electrochemical response (as evident where microgram quantities of graphene are deposited onto the electrode surface in our case). A schematic representation of the transition between these two extremes, where the basal dominant single layered graphene materialises into the edge dominated graphite response, is depicted in Fig. 8 which is representative of the modification of a fast electron transfer underlying electrode towards a typical outer-sphere redox probe. In addition to this, Fig. 9 depicts the observable change in the composition of the electrode surface and the resultant electrochemical responses expected, where Fig. 9A is that of a HOPG electrode assumed to possess fast electron transfer kinetics, Fig. 9B after incomplete coverage of single-layered graphene, Fig. 9C after complete single-layer coverage of graphene (represented via the dotted line in Fig. 8 at the interface between the two working zones), and Fig. 9D where increased graphene content leads to the coalescing of graphene sheets into double-, few-, and multi- layered graphene (vizgraphite): note that until layering begins there is a continual decrease in the electrochemical response owing to graphene blocking electron transfer at the underlying electrode surface (Zone I in Fig. 8), and conversely when multiple layers materialise an improvement in the electrochemical response is observed owing to increased electron transfer at the electrochemically active edge plane sites (Zone II in Fig. 8). Note that a ‘Zone III’ will likely exist if the graphene exhibits ‘thin-layer’ behaviour where the peak-to-peak separation will become less than 60 mV, however recent literature has shown that for the type of commercially available graphene utilised and the analytes studies, graphene does not suffer from this thin-layer behaviour,49 however, there may be exceptions.
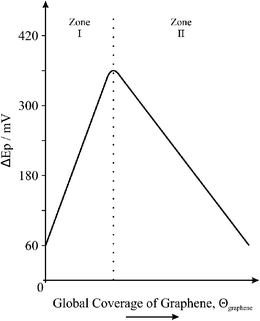 | ||
| Fig. 8 The effect of the global coverage of graphene, Θgraphene, on the heterogeneous electron transfer rate/kinetics, indicating two distinctive regions that are commonly encountered (versus peak-to-peak separation, ΔEp, in this case to simulate a standard redox probe where large ΔEp values are indicative of slow electron transfer): note that in this case the underlying electrode substrate is assumed to possess fast electron transfer rate kinetics and the redox probe is a simple outer-sphere species. | ||
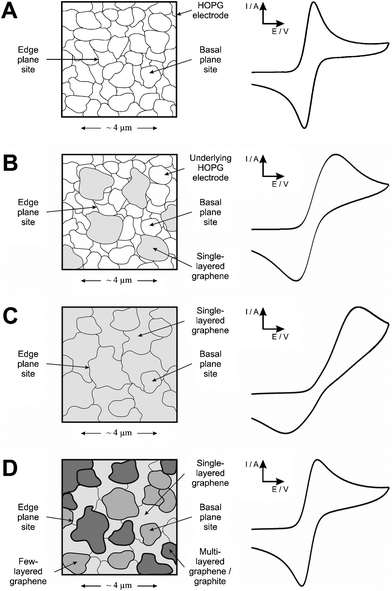 | ||
| Fig. 9 Schematic representation of the effect on the voltammetric performance resulting from differing coverages of graphene using a simple outer-sphere electron transfer redox probe. (A) represents an unmodified HOPG electrode surface where fast electron transfer kinetics are observable, (B) after modification with graphene leading to incomplete coverage where reduced electron transfer rates occur, (C) after modification with graphene leading to complete single layer coverage where due to the large basal content of graphene (in contrast to edge plane) poor electrochemical activity is observed where electron transfer is effectively blocked, and (D) after continual modification with graphene leading to layered structures with increased edge plane sites available (origin of fast electron transfer) and thus an improvement in the electrochemical response is observed. | ||
In the above case an underlying electrode with fast electron transfer is modified with graphene (vizEPPG electrode). To investigate this further we next explore the modification of a BPPG electrode with nanogram quantities of graphene towards the inner-sphere redox probe 1 mM potassium ferrocyanide (II). As depicted in Fig. 10 the addition of 5 and 40 ng of graphene upon the BPPG electrode surface results in the increased peak-to-peak separation of the redox probe indicating slower heterogeneous electron transfer at graphene over that of the unmodified BPPG electrode, indicating a partially blocked electrode (Zone I—see above). Interestingly however, with further additions of graphene, within the microgram region (0.5, 3.0 and 5.0 μg), the voltammetric response continues to exhibit increasing peak-to-peak separations before the surface becomes completely blocked, which is distinctly different to that observed above in Fig. 7A. Thus there is contrasting behaviour between modifying an EPPG and a BPPG electrode with graphene.
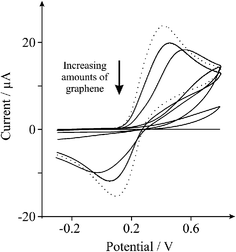 | ||
| Fig. 10 Cyclic voltammetric profiles recorded utilising 1 mM potassium ferrocyanide (II) in 1 M KCl, obtained using a BPPG electrode (dotted line) with the addition of 5 and 40 ng, and 0.5, 3.0 and 5.0 μg of graphene (solid lines). Scan rate: 100 mVs−1 (vs.SCE). | ||
We next turn to exploring the orientation of the immobilised graphene on both EPPG and BPPG surfaces viaScanning Electron Microscopy (SEM). Fig. 11A–C depicts SEM images of the EPPG surface before and after modification with graphene which exhibits coalesced graphene folding over edge plane sites; this potentially explains the blocking effect observed in Fig. 2A, 3, 4A, and 5 as discussed above. When further graphene is immobilised onto the electrode surface this then realigns in orientation with the edge plane sites of the underlying electrode resulting in vertically aligned graphene and hence a beneficial increase in the electrochemical response is observed due to the increment in the proportion of edge plane sites accessible for electron transfer; this response is shown schematically in Fig. 12. In this model we assume that the graphene will adopt a similar architecture to that of the underlying electrode since graphene has a distributed electron density of the planar–basal site (π–π) which will be disturbed by the high electron density of the underlying edge sites of graphene (the EPPG) such that it effectively ‘aligns’ with the underlying electrode surface as this arrangement reflects the lowest energy settlement. Due though to the high number of graphene sheets on the EPPG this will force the graphene sheets to stack (as a continuation of the edge planes) in parallel to each other in order to fit the limited space of the EPPG surface.
 | ||
| Fig. 11 SEM images of an unpolished EPPG electrode before (A) and after modification with low (B) and high (C) coverage's of graphene, and additionally an unpolished BPPG electrode before (D) and after modification with low (E) and high (F) coverage's of graphene. | ||
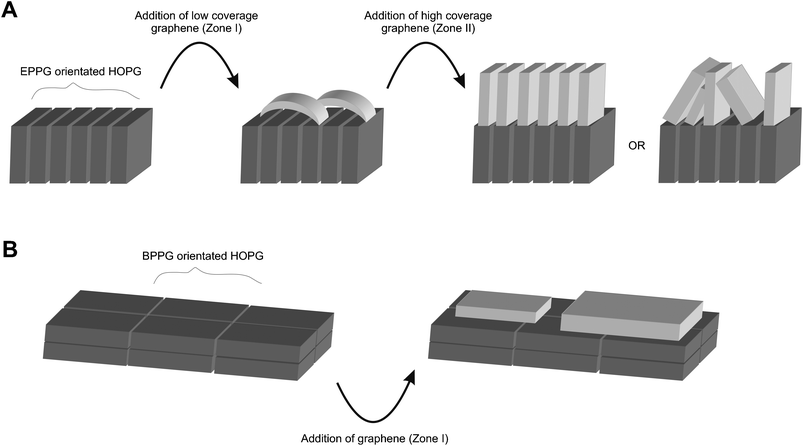 | ||
| Fig. 12 A schematic representation of the differentiating orientations of graphene sheets encountered depending upon the architecture of the underlying substrate, where variations occur between low and high coverage's in the case of modifying an EPPG electrode(A), and the modification of a BPPG electrode(B) results in a singular orientation. | ||
Fig. 11D–F depicts SEM images of a basal plane surface before and after modification with graphene, which are distinctively different to those observed above, indicating a different orientation of graphene on the electrode surface. In this case, the immobilised graphene follows the same architecture presented on the BPPG sheets meaning that the graphene will stack planar on the BPPG due to π–π stacking. This explains the experimental result observed in Fig. 10 and is shown schematically in Fig. 12. Note however, it might be likely that when graphene is further deposited an improvement in the electrochemical response might be observed at extremely high graphene coverages which would be a highly porous and rough surface with poor reproducibility which is effectively approaching that of a graphite modified electrode, in this case the electrochemical response is expected to reach that of Zone II.
Last, the electron distribution in graphene was probed utilising computational DFT calculations. Five models were used to represent increasingly large symmetric flakes of graphene which have m rings in the top row and n rows of rings along each diagonal edge to give an m × n sheet: 2 × 2 sheet (coronene), 3 × 3 sheet, 4 × 4 sheet, 5 × 5 sheet and 6 × 6 sheet. The surface-area ratio of the basal plane to the edge plane increases from 1.34 (for coronene) to 3.96 for the 6 × 6 sheet of graphene. Although these surface-area ratios are significantly lower than those found in the experimental samples, this series of models can still provide valuable insights. As can be seen in Fig. 13, the electron density in both the highest-occupied molecular orbitals (HOMO) and lowest-unoccupied molecular orbitals (LUMO) is concentrated around the edge of the graphene sheets, even in flakes as small as the 3 × 3 sheet shown in Fig. 13B. The side view shows that any electron density concentrated in the basal region is less than that concentrated at the edges. The probability that the electrons will be concentrated around the edge of the graphene flake increases as the size of the sheet increases (Fig. 13C and also for the larger models, although not shown). Thus, in graphene, the electrons that have the highest energies, and are therefore the most likely to transfer, have a higher probability of being concentrated in the edge plane than in the central basal plane region, which is consistent with the experimental results and fundamental understanding regarding the electron transfer sites of graphitic materials. In contrast, in coronene (shown in Fig. 13A), the electron density is delocalised across the whole molecule, which may decrease the availability of the high-energy electrons and concomitantly decrease the electron transfer rate for this molecule.
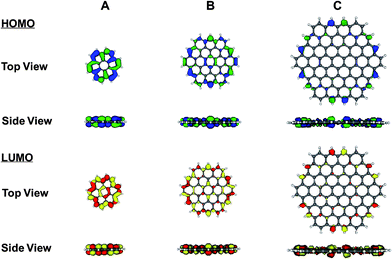 | ||
| Fig. 13 Electron density distribution of electrons in the HOMO (shown in blue and green) and the LUMO (shown in red and yellow) of a (A) 2×2 sheet (coronene), (B) 3×3 sheet, and (C) 4×4 sheet of graphene. Due to the symmetry of the sheets, the HOMO and HOMO–1 are degenerate in energy and both are displayed simultaneously. Similarly, the LUMO and LUMO + 1 are degenerate in energy and both are displayed simultaneously in these figures. In each case, the 0.035 a.u. surface is shown. All molecular orbital diagrams were visualised using MOLEKEL 4.2.51,52 | ||
Conclusions
We have reported for the first time the effect of coverage when utilising graphene as an electrode material and compared the physicoelectrochemical electron transfer nature of graphene to that of graphite utilising a range of electrochemically well characterised analytes. We find that the use of graphene in electrochemical sensing applications is questionable, where interestingly we observe that graphene blocks underlying electrode activity, exhibiting slow heterogeneous electron transfer kinetics owing to its large basal and low edge plane content. Insights from DFT calculations confirm that the electron density is concentrated around the edge of the graphene indicating that the edge to basal ratio is critical for graphene (and graphitic materials alike) and as it has a low proportion of edge plane sites graphene resultantly exhibits poor electrochemical responses towards various analytes.We have demonstrated that there are two essential coverage regions for graphene (for the case where the underlying electrode exhibits fast electron transfer, such as in the case of an EPPG electrode), where in ‘Zone I’ low global coverage of graphene, Θgraphene, immobilised onto an electrode substrate exhibits slow heterogeneous electron transfer owing to a large basal plane content. Conversely in ‘Zone II’, equating to high coverage of graphene, fast heterogeneous electron transfer is observed as layering leads to increased edge plane content. Interestingly these Zones are not observed when the underlying electrode is switched from an EPPG to a BPPG electrode and complementary SEM analysis provides insights into this differing behaviour indicating that the orientation of the graphene and the underlying supporting electrode is of key importance.
In our work we are the first to show, via the quantification of heterogeneous electron transfer, that the use of graphene is not so beneficial, as commonly (mis)reported in the literature, as an “electrocatalytic” electrode material in terms of the low proportion of electron transfer sites it possesses (edge plane content) resulting in slow electrode kinetics and consequently (for these select analytes) we conclude that graphite is ultimately far superior in terms of electrochemical activity and electron transfer kinetics, thus in the fabrication of sensing devices we strongly suggest the use of graphite be considered over that of graphene, or at least control experiments be diligently reported. Note however, that this is purely from the perspective of electrode kinetics (bearing in mind that fast heterogeneous electron transfer is not the only criteria for a good electrode material and inversely in some cases slow electron transfer may be considered an advantage), thus in addition to such select sensing applications where this is the case we note that graphene has huge potential in energy storage and generation applications.5,50
Acknowledgements
The authors thank Maria Gómez-Mingot and Jesús Iniesta (Alicante University, Spain) for aiding in the acquirement of SEM images.References
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS.
- D. A. C. Brownson and C. E. Banks, Analyst, 2010, 135, 2768 RSC.
- M. Pumera, Chem. Rec., 2009, 9, 211 CrossRef CAS.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef CAS.
- D. A. C. Brownson, D. K. Kampouris and C. E. Banks, J. Power Sources, 2011, 196, 4873 CrossRef CAS.
- S-M. Paek, E. Yoo and I. Honma, Nano Lett., 2009, 9, 72 CrossRef CAS.
- H. Wang, Q. Hao, X. Yang, L. Lu and X. Wang, Electrochem. Commun., 2009, 11, 1158 CrossRef CAS.
- C. Liu, S. Alwarappan, Z. Chen, X. Kong and C. Z. Li, Biosens. Bioelectron., 2010, 25, 1829 CrossRef CAS.
- R. I. Jafri, N. Rajalakshmi and S. Ramaprabhu, J. Mater. Chem., 2010, 20, 7114 RSC.
- X. Kang, J. Wang, H. Wu, J. Liu, I. A. Aksay and Y. Lin, Talanta, 2010, 81, 754 CrossRef CAS.
- Y-R. Kim, S. Bong, Y. J. Kang, Y. Yang, R. K. Mahajan, J. S. Kim and H. Kim, Biosens. Bioelectron., 2010, 25, 2366 CrossRef CAS.
- X. Kang, J. Wang, H. Wu, I. A. Aksay, J. Liu and Y. Lin, Biosens. Bioelectron., 2009, 25, 901 CrossRef CAS.
- Y. Wang, Y. Wan and D. Zhang, Electrochem. Commun., 2010, 12, 187 CrossRef CAS.
- J-F. Wu, M. Q. Xu and G. C. Zhao, Electrochem. Commun., 2010, 12, 175 CrossRef CAS.
- M. S. Goh and M. Pumera, Anal. Chem., 2010, 82, 8367 CrossRef CAS.
- D. A. C. Brownson and C. E. Banks, Electrochem. Commun., 2011, 13, 111 CrossRef CAS.
- D. A. C. Brownson, J. P. Metters, D. K. Kampouris and C. E. Banks, Electroanalysis, 2011, 23, 894 CrossRef CAS.
- D. A. C. Brownson and C. E. Banks, Analyst, 2011, 136, 2084 RSC.
- D. K. Kampouris and C. E. Banks, Chem. Commun., 2010, 46, 8986 RSC.
- M. A. Edwards, P. Bertoncello and P. R. Unwin, J. Phys. Chem. C, 2009, 113, 9218 CAS.
- A. Dato, V. Radmilovic, Z. Lee, J. Phillips and M. Frenklach, Nano Lett., 2008, 8, 2012 CrossRef CAS.
- A. Dato, Z. Lee, K. J. Jeon, R. Erni, V. Radmilovic, T. J. Richardson and M. Frenklach, Chem. Commun., 2009, 6095 RSC.
- Z. Lee, K. J. Jeon, A. Dato, R. Erni, T. J. Richardson, M. Frenklach and V. Radmilovic, Nano Lett., 2009, 9, 3365 CrossRef CAS.
- www.graphene-supermarket.com .
- F. Neese, An ab initio, density functional and semi-empirical program package–Version 2.8, ( 1999–2011) ORCA, University of Bonn Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623 CrossRef CAS.
- X. Ji, C. E. Banks, A. Crossley and R. G. Compton, ChemPhysChem, 2006, 7, 1337 CrossRef CAS.
- C. E. Banks, R. R. Moore, T. J. Davies and R. G. Compton, Chem. Commun., 2004, 1804 RSC.
- A. S. Adekunle and K. I. Ozoemena, Electrochim. Acta, 2008, 53, 5774 CrossRef CAS.
- T. J. Davies, M. E. Hyde and R. G. Compton, Angew. Chem., Int. Ed., 2005, 44, 5121 CrossRef CAS.
- W-J. Lin, C. S. Liao, J. H. Jhang and Y-C. Tsai, Electrochem. Commun., 2009, 11, 2153 CrossRef CAS.
- C. E. Banks, T. J. Davies, G. G. Wildgoose and R. G. Compton, Chem. Commun., 2005, 829 RSC.
- C. E. Banks and R. G. Compton, Analyst, 2006, 131, 15 RSC.
- C. E. Banks and R. G. Compton, Analyst, 2005, 130, 1232 RSC.
- T. J. Davies, R. R. Moore, C. E. Banks and R. G. Compton, J. Electroanal. Chem., 2004, 574, 123 CrossRef CAS.
- M. C. Henstridge, L. Shao, G. G. Wildgoose, R. G. Compton, G. Tobias and M. L. H. Green, Electroanalysis, 2008, 20, 498 CrossRef CAS.
- F. Wantz, C. E. Banks and R. G. Compton, Electroanalysis, 2005, 17, 1529 CrossRef CAS.
- C. Amatore, J. M. Saveant and D. Tessier, J. Electroanal. Chem., 1983, 147, 39 CrossRef CAS.
- T. J. Davies, E. R. Lowe, S. J. Wilkins and R. G. Compton, ChemPhysChem, 2005, 6, 1340 CrossRef CAS.
- R. S. Nicholson, Anal. Chem., 1965, 37, 1351 CrossRef CAS.
- C. E. Banks, R. G. Compton, A. C. Fisher and I. E. Henley, Phys. Chem. Chem. Phys., 2004, 6, 3147 RSC.
- Y. Wang, J. G. Limon-Petersen and R. G. Compton, J. Electroanal. Chem., 2011, 652, 13 CrossRef CAS.
- R. L. McCreery, Chem. Rev., 2008, 108, 2646 CrossRef CAS.
- T. J. Davies and R. G. Compton, J. Electroanal. Chem., 2005, 585, 63 CrossRef CAS.
- T. J. Davies, S. Ward-Jones, C. E. Banks, J. Del Campo, R. Mas, F. X. Muñoz and R. G. Compton, J. Electroanal. Chem., 2005, 585, 51 CrossRef CAS.
- T. J. Davies, C. E. Banks and R. G. Compton, J. Solid State Electrochem., 2005, 9, 797 CrossRef CAS.
- P. M. Hallam and C. E. Banks, Electrochem. Commun., 2011, 13, 8 CrossRef CAS.
- D. A. C. Brownson and C. E. Banks, Chem. Commun., 2012 10.1039/c1cc11276g.
- P. Flukiger, H. P. Luthi, S. Portmann and J. Weber, Swiss Center for Scientific Computing, ( 2000–2009), Manno (Switzerland) Search PubMed.
- S. Portmann and H. P. Lüthi, Chimia, 2000, 54, 766 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1ra00393c |
| This journal is © The Royal Society of Chemistry 2011 |