Synthetically useful carbon–carbon and carbon–sulphur bond construction mediated by carbon- and sulphur-centred radicals in water and aqueous media
Al
Postigo
*
Facultad de Farmacia y Bioquímica, Universidad de Buenos Aires, Junín 934, Buenos Aires, Argentina. E-mail: alberto.postigo@ub.edu.ar
First published on 26th July 2011
Abstract
This account is focused on carbon–carbon, and carbon–sulphur bond-forming reactions through the use of carbon- and sulphur-centred radicals in water, respectively. It intends to show the scope and applications of these types of radicals with a synthetic goal, thus helping the radical synthetic chemist to grasp the fundamental aspects of the syntheses of a wide array of organic compounds through radical methods in environmentally friendly media. This account excludes the syntheses of organic substrates with the aid of organometallic/metal-centred radicals such as other Group IV-radicals and transition-metal radicals, and the important class of radical reductions currently accomplished in water using different metallic reducing species. The array of substrates synthesized through the present method encompasses intermolecular and intramolecular radical carbon–carbon bond formation reactions in water, and radical carbon–sulphur bond formation reactions. Among the radical carbon–carbon bond formation reactions, special attention is devoted to atom-transfer radical reactions, radical addition reactions to olefins, radical addition to carbon–nitrogen double bonds, the carboaminohydroxylation of alkenes, and the analogues of Knovonoegel reactions, the synthesis of functionalized enol ethers, thr carboazidation and azidation, and ynol formation, and radical cyclization reactions in water.
1. Introduction
The chemistry of free radicals has undergone a massive renaissance over the past thirty years. The real burst in synthetic applications arose from the use of trialkyltin and triaryltin hydrides as radical-chain carriers. However, the toxicity of the tin reagents, coupled with the difficulty in separation of their by-products from the desired reaction products, meant that they were never acceptable to the pharmaceutical industry. Although improved methods of separation and operation have been developed, there is still a reluctance to use toxic tin reagents. This is unfortunate, since the properties of free radicals—e.g. lack of solvation (useful in assembling congested quaternary carbons) and predictable kinetics regardless of the reaction solvent (useful for predicting the relative speed of desired vs. side-reactions) give them a unique advantage in certain synthetic manoeuvres. Recently, silicon hydrides, in combination with sulphides have been used in water as efficient chain carriers, with radical chain carrying rates equalling those of tin hydrides. However, this important class of non-carbon-centred radicals is not the subject of this review article.The utilisation of solvents that have a reduced impact on the environment and serve as alternatives to the currently used volatile and/or flammable organic solvents, are considered central to designing a specific synthetic protocol. Particularly, in large-scale and industrial procedures, harmful solvent waste minimization and disposal are critical in selecting synthetic strategies in order to reduce costs and pollution. Thus the number of research articles on radical reactions in water has awakened the need for guiding chemists toward further research and discoveries. In this sense there has recently been an account discussing the different properties that alternative green solvents have, recognizing whether a solvent is actually green, and finding easily-removable polar aprotic solvents and eliminating distillation.1a
In radical chain processes, the initial radicals are generated by some form of initiation. In organic solvents, the most popular initiator is 2′,2′-azobisisobutyronitrile (AIBN), with a half-life of 1 h at 81 °C, generating the incipient radicals that commence the radical chain reaction. Other azo-compounds are used from time to time as is the thermal decomposition of di-tertbutyl peroxide depending on the reaction conditions. Triethylborane (Et3B) in the presence of very small amounts of oxygen is an excellent initiator for low temperature reactions (down to −78 °C). Also air-initiated reactions have recently been reported in aqueous and neat mixtures. However, in water, the method of radical initiation varies, and several studies have been undertaken to assess the best methodology. In the last decade, Et3B/dioxygen has also been used as radical initiator in water.
Following the progressive exploration of radical reactions in non-conventional media, water has been adopted as the solvent of choice as advantages such as rate acceleration, stereoselectivity, and regioselectivity have been observed when radical reactions are performed in this latter medium as compared to organic solvents. However, the following considerations should be made clear:
Solvent polarity can have tremendous effects on the kinetics of reactions involving charged species in solution. On the other hand, reactions of neutral radicals are less sensitive to solvent polarity effects, mainly because charged species are not involved, and there is not a significant change in dipole moment in the progression from reactants to transition state. However, other solvent properties such as viscosity or internal pressure can influence the rate of certain radical reactions; such solvent effects are much more difficult to detect in polar reactions because they are masked by the overwhelming effect of solvent polarity.1b
For example, solvent viscosity can affect the rate and product distribution when radical caged-pairs (geminate or diffusive) are involved. Internal pressure can influence the rate if there is a difference in the volume of the reactants compared to the transition state (ΔVact ≠ 0) and can influence the relative rate of some radical reactions. However, these solvent effects are generally small, with changes in rate or product distribution not much greater than an order of magnitude. Consequently, instances where solvent dramatically affects the rate or selectivity of reactions involving neutral radicals are rare and therefore noteworthy.
The classic example of a significant solvent effect in a radical reaction involves the free radical chlorinations of alkanes conducted in benzene. The chain-carrying chlorine atom forms a complex with benzene, lowering its reactivity and increasing its selectivity (by nearly 2 orders of magnitude) in hydrogen atom abstractions. A more recent example of a significant solvent effect was reported by Ingold and co-workers, who found that rate constants for hydrogen atom abstractions from phenols were reduced in solvents where the phenol was stabilized by hydrogen bonding.1c In this case, it was the reactivity of the substrate, not the radical, that was diminished as a result of a solute/solvent interaction.
In the last few years, solvent effects have been extensively investigated by Ingold and co-workers.2 This fundamental work can be summarized briefly as follows:
(i) There are generally no solvent effects on the rates of hydrogen atom abstraction from a C–H bond;
(ii) There may be solvent effects on the rates of unimolecular radical scission reactions but these are generally fairly small;
(iii) There are large solvent effects on the rates of hydrogen atom abstraction from O–H bonds (and to a lesser extend from N–H bonds).3–6
Point (i) means that radical reactivities, insofar as hydrogen abstraction (and addition) reactions are concerned, are not influenced by the solvent. Point (iii) means that O–H containing substrates (e.g., phenols, hydroperoxides, etc.) have their reactivities towards radicals influenced by the solvent. Therefore, it was predicted that the KSE (kinetic secondary isotope effect) should be dependent on the ability of XOH to participate as a hydrogen bond donor to HBA (hydrogen donor) solvents but should be independent of the reactivity or nature of the radical which does the hydrogen atom abstraction. This prediction, which is the first new and unifying principle for organic free radical chemistry to have been proposed in the last forty years, has been dramatically confirmed for hydrogen abstraction from phenol by the highly reactive cumyloxyl radical, PhCMe2O,5,6 and by the very unreactive diphenyl picrylhydrazyl radical, DPPH, Ph2N•NC6H2(NO2)3.
However large or small solvents effects may be on the rates of radical reactions, radical reactivity in water has proved a source of facile and unexpected organic transformations. In the present work, we shall embark on presenting the scope of radical carbon–carbon and carbon–sulphur bond forming reactions in water, with an emphasis on the regio- and stereoselectivity achieved in this medium in comparison to organic solvents.
2. Radical carbon–carbon bond formation in water
2.1. Radical carbon–carbon bond formation from olefins in water
The fastest rate constants ever reported for carbon-centred radical reactions were determined by Ingold and Lusztyk in water as the sole solvent.7 Aryl radicals generated from sodium 4-iodophenylsulfonate by laser flash photolysis reacted with non-activated olefins at rates of k ≈ 2–3 × 107 M−1 s−1, and the authors pointed out that the fast and selective aryl radical addition could be due to the aqueous solvent.For comparison, rate constants as low as k ≈ 5 × 105 M−1 s−1 were derived in earlier experiments conducted in tetrachlorocarbon.8
A key element to achieve selectivity in reactions of aryl radicals appears to be the use of water as the solvent or at least as co-solvent.7
Ingold and Dolbier conducted a laser flash photolysis (LFP) study9 on the rates of perfluoroalkyl radical addition to double bonds and found that the rate of addition in water is significantly larger than in perfluorocarbon solvents.
The authors9 studied the addition of water-soluble radicals •RfSO3− to water-soluble alkenes of the type CH2=CRCO2Na or other carboxylate-substituted terminal alkenes. They postulated that the observed rate enhancements in water most probably reflect the greater ability of this polar solvent to stabilize the polar transition state for addition of •RfSO3− to the alkenes, such as that depicted in Fig. 1.
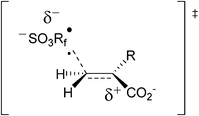 | ||
| Fig. 1 Coulombic effects postulated on the transition state for the radical addition of •RfSO3− onto alkenes. | ||
The observed rate enhancements are all the more remarkable in view of the Coulombic repulsion that must be met in a transition state such as that depicted in Fig. 1, involving two negatively charged ions. It was recognized as early as 1922 by Brønsted that for “ions of the same sign the repulsive forces will tend to keep them apart”.
A general expectation is that the addition reactions between the negatively charged radical and negatively charged alkene substrates would be inhibited by electrostatic repulsion. However, the presence of a polar solvent or a medium of increased ionic strength should at least partially ameliorate the inherently detrimental electrostatic effects present in a reaction between two negatively charged ions and, hence, facilitate such reactions, mainly by stabilization from hydrogen-bonds and charge dispersion. The authors9 also postulate the presence of ionic strength effects.
Considering the dilute solutions used in the LFP experiments the authors arrive at the conclusion that solvent effects provide the simplest and most reasonable explanation for the observed rate constant differences. If not for the negative impact of Coulombic repulsion in the transition state, these rate enhancements would most certainly be much larger.
Dolbier et al.10 have found that perfluorinated radicals were much more reactive than their hydrocarbon counterparts in addition to normal, electron rich alkenes such as 1-hexene (40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times more reactive) in organic solvents, and that H transfer from (Me3Si)3SiH to a perfluoro-n-alkyl radical such as n-C7F15CH2CH•C4H9 was 110 times more rapid than the analogous hydrocarbon radicals eqn (21).
000 times more reactive) in organic solvents, and that H transfer from (Me3Si)3SiH to a perfluoro-n-alkyl radical such as n-C7F15CH2CH•C4H9 was 110 times more rapid than the analogous hydrocarbon radicals eqn (21).
Barata-Vallejo and Postigo11 have reported the radical perfluoroalkylation of organic solvent-soluble alkenes in water, and found that the relative rates of radical addition/reduction in water were higher than those reported in organic solvents, for consecutive reactions.
According to what has been observed and measured by Dolbier et al.10, in benzene-d6, the ratio of products [1]/[2] eqn (1) should equal the ratio of the rate constants for addition (of perfluorinated heptyl radical to 1-hexene) and the rate constant for H abstraction from (Me3Si)3SiH times the ratio of concentrations of alkene and silane.
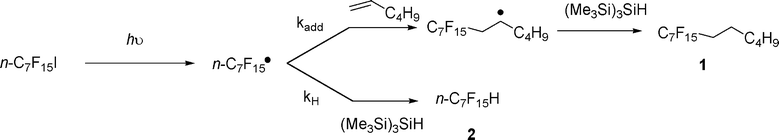 | (1) |
According to the experimental conditions reported by Barata-Vallejo and Postigo,11 by employing eqn (2), the authors would obtain a theoretical ratio of perfluoroalkylated alkane to reduced perfluoroalkane of ca.1.3, which is not completely in agreement with the unobserved reduced perfluoroalkanes in their reaction systems (i.e.: CHF2(CF2)4CF3, when iodoperfluorohexane is employed).11
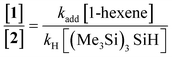 | (2) |
The electrophilicity of Rf• radicals is the dominant factor giving rise to their high reactivity. The stronger the carbon–carbon bond which forms when Rf•versus R• radicals add to an alkene is a driving force for the radical addition (the greater exothermicity of the Rf• radical addition is expected to lower the activation energy).12 It has been observed in organic solvents, that the rates of addition of Rf• radicals to alkenes correlate with the alkene IP (ionization potential) (which reflects the HOMO energies).13 Indeed, the major transition state orbital interaction for the addition of the highly electrophilic Rf• radical to an alkene is that between the SOMO of the radical and the HOMO of the alkene. Thus, the rates of Rf• radical addition to electron deficient alkenes are slower than those of addition to electron rich alkenes (as observed in organic solvents). From the results in water,11 however, it becomes apparent, that in water the reactivity for both electron rich and electron deficient alkenes towards Rf• radical addition could be comparable. In order to clarify this subtle aspect of the reaction in water, the authors11 undertook a set of experiments designed to compare the ratios of (kH/kadd)1-hexene and (kH/kadd)acrylonitrile for the addition reaction of n-C6F13I to the electron rich 1-hexene and the electron deficient acrylonitrile, respectively. These ratios of rate constants are obtained by plotting [n-C6F13H]/[C6F13–C6H13] vs. [(Me3Si)3SiH]/[1-hexene] and [n-C6F13H]/[C6F13–CH2CH2CN] vs. [(Me3Si)3SiH]/[acrylonitrile], respectively, when the reactions are initiated thermally, by using incremental amounts of (Me3Si)3SiH, and keeping the alkene and n-C6F13I concentrations constant. They obtained slopes for both plots equal to 1.55 ± 0.09 (r2 = 0.998) and 1.88 ± 0.19 (r2 = 0.989) for (kH/kadd)1-hexene and (kH/kadd)acrylonitrile respectively. This seems to indicate that the reactivities of electron rich and electron deficient alkenes towards Rf• radicals in water are levelled off.13
A tandem radical addition–oxidation sequence which converts alkenyl silanes into ketones has been elegantly described by Oshima et al.14
When 2-silyl-1-alkenes (carbonyl synthons) are made to react with an excess of Et3B (2 equiv.) in air, methyl ketones are afforded as major products as shown in Scheme 1 below. Water was found to be an excellent solvent. The addition of ammonium chloride is important as without it, the yields drop substantially.
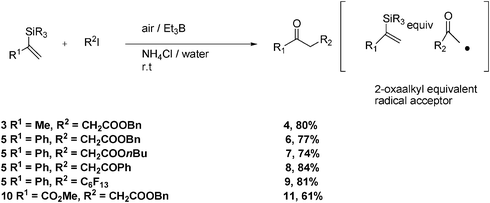
When the methyl-substituted alkenyl silane 3 is allowed to react with benzyl-2-iodoacetate under the reaction conditions described above, compound 4 is obtained in 80% yield (Scheme 1). Analogously, from phenyl-substituted alkenyl silane 5 and benzyl-2-iodoacetate, compound 6 is obtained in 77% yield. When butyl-2-iodoacetate and 2-iodo-1-phenylethanone react with 5, compounds 7 and 8 are obtained in 74% and 84% yields, respectively. When iodoperfluoro-n-hexane is allowed to react with 5, compound 9 is obtained in 81% isolated yield. The reaction of methylformate-substituted alkenyl silane 10 with benzyl-2-iodoacetate affords product 11 in 61% yield (Scheme 1).
The authors15 propose a plausible reaction mechanism such as that depicted in Scheme 2.
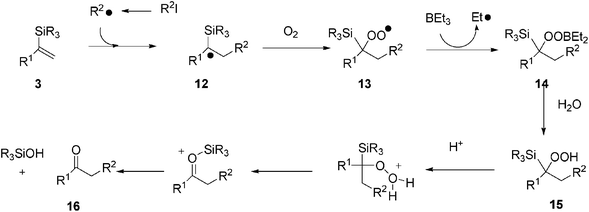 | ||
| Scheme 2 Reaction mechanism of Et3B-induced synthesis of ketones from alkenylsilanes and iodoalkanes in water. | ||
Addition of a radical arising from benzyl-2-iodoacetate to, for instance, alkenylsilane 3 affords an α-silyl radical 12 which then reacts with dioxygen, to afford peroxyl radical 13. The reaction of radical 13 with Et3B generates peroxyborane 14. Hydroperoxide 15, derived from the hydrolysis of 14, is eventually converted to carbonyl compound 16, through migration of the silyl group to the internal oxygen atom. The ethyl radical which results from the reaction 13 and 14, regenerates an alkyl radical from RI (Scheme 2).
2.2. Radical carbon–carbon bond formation from imines in water
The carbon–nitrogen double bond of imine derivatives has emerged as a radical acceptor, and thus numerous synthetically useful carbon–carbon bond-forming reactions are available.Naito et al.16 utilized oximes, oxime ethers, hydrazones, and nitrones as radical acceptors for construction of new carbon–carbon bonds.
Thus, when the glyoxylic oxime ether 17 is added to iso-propyl iodide in a methanol/water mixture with Et3B as initiator, a quantitative conversion to 18 is obtained, see eqn (3).
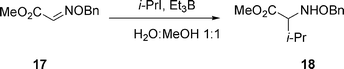 | (3) |
The conventional condensation of glyoxylic acid hydrate 19 with benzyloxyamine hydrochloride proceeds smoothly in water. Subsequently, alkyl iodide (RI) and Et3B are added to the reaction vessel to afford excellent yields of α-amino acid derivatives 20a–d (eqn (4)) after the purification. It should be noted that the one-pot reactions in water are much more effective compared to the reactions carried out in organic solvents such as toluene and methylene chloride.16
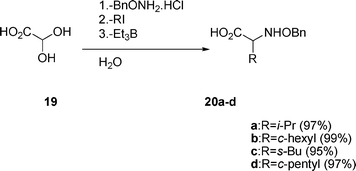 | (4) |
Other primary and secondary alkyl radical additions have also been attempted successfully on glyoxylic oxime ethers. Scheme 3 depicts the scope of glyoxylic oxime ethers as excellent radical acceptors in water/methanol mixtures.17
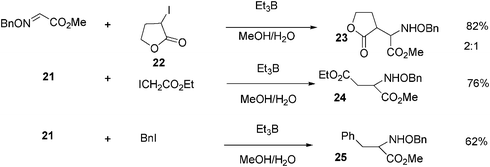 | ||
| Scheme 3 Reaction of glyoxylic oxime ethers with iodocompounds in water. | ||
When glyoxylic oxime ether 21 reacts in a methanol/water mixture with dihydro-3-iodofuran-2(3H)-one 22, in the presence of Et3B, product 23 is obtained in 82% yield, with a diastereomeric ratio equal to 2:1 (Scheme 3). Iodoethylacetate, under the same reaction conditions affords product 24 in 76% yield, while benzyl iodide, affords product 25 in 62% yield.17
Ketimines have also been used as alkyl radical acceptors in water to construct new carbon–carbon bonds.18 In particular, ketimines having N-heteroatom substituents stabilizing the intermediate aminyl radical or the N-alkyl group, have been used in the intermolecular radical reaction.
The aqueous-medium alkyl-radical addition to hydrazine 26 proceeds efficiently in the presence of alkyl iodide with Et3B/dioxygen as initiator (eqn (5)) to afford 27 The aqueous-medium reaction of isatin hydrazine 28 affords the product 29, see eqn (6).
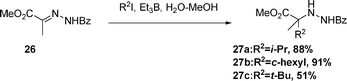 | (5) |
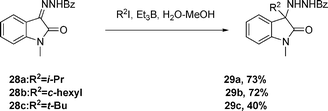 | (6) |
2.3. Radical atom transfer reactions for intermolecular carbon–carbon bond formation in water
Radical atom transfer reactions can encompass the distinct transfer of atoms or group of atoms. Halogen atoms or azide moieties can be transferred during intermolecular radical atom transfer reactions in water.Halogen atom-transfer (HAT) has been extensively studied and widely used in organic synthesis. The addition of a carbon–halogen bond across a double bond was pioneered by Kharasch19 (Scheme 4) and provides new carbon–carbon and carbon–halogen bonds in a single operation. The choice of the halogen that transfers in the reaction is crucial for the success of atom-transfer additions.
When triethylborane (Et3B) is added to a suspension of ethyl bromoacetate 30 and 1-octene 31 in water under argon (introducing air slowly), 4-bromodecanoate 32 is obtained in 80% yield, see eqn (7).15,20 The same authors also report that other bromides besides ethyl bromoacetate undergo radical addition. Bromomalonate, and bromoacetonitrile give excellent results for bromine atom transfer products.
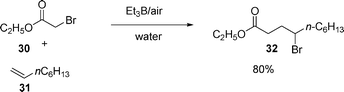 | (7) |
As has been explained above, radical atom transfer reactions can effectively be initiated by Et3B in water. Since radical carboazidations are generated via the initial transfer of N iodine atom or xanthate group, it becomes reasonable that these reactions also be initiated by Et3B in water.
The Et3B-induced reaction of alkene 31 with iodoacetate ethyl ester and phenylsulfonyl azide in water yields azide 33 in 85% yield (eqn (8)).21 Cycloalkene 34, under the same reaction conditions, affords 35 in 90% yield eqn (9).
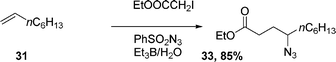 | (8) |
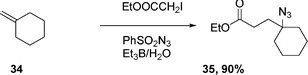 | (9) |
2.4. The Knoevenagel reaction in aqueous media
Carbon–carbon bond formation through a free radical pathway has led to a variety of useful applications in organic synthesis (vide supra). Thus, a series of functionalized reactions of ylidenemalonitriles prepared in situ by reaction of carbonyl compounds and malononitrile mediated by Et3B in the presence of ammonium acetate in aqueous media (biphasic with Et2O) have been attempted to render compounds 40 in 80–96% yields with an array of carbonyl compounds 36a–c, eqn (10).22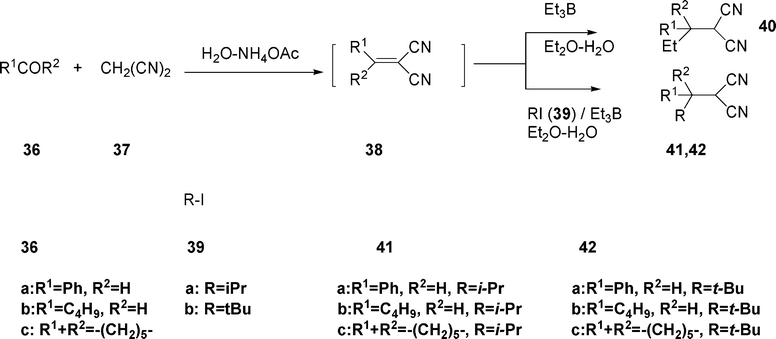 | (10) |
Similarly, high yields of 41 and 42 were obtained when 38 was reacted with Et3B (3 equiv.) and iso-propyl iodide or tert-butyl iodide under similar reaction conditions. In addition to malononitrile 37, dimethylmalonate was employed.
2.5. Carboaminohydroxylation of olefins in water
Ingold and Lusztyk pointed out that aqueous solvents are a critical element for controlled reactions of aryl radicals.7When aryldiazonium salts 43, tetramethylpiperidine oxyl radical (TEMPO, a persistent radical) 44, and various olefins 45 are made to react in DMSO–water, adduct 46 is afforded (Scheme 5). In all cases, the yields of the reactions demonstrate that the method is synthetically useful.23
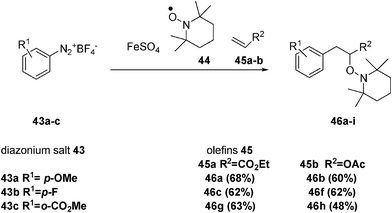
Interestingly, the reactions, including ethyl acrylate (45a) (Scheme 5, column 2), do not generally give the best yields, although they are favoured in two ways: first by the fast addition of the aryl radical to the olefin and second by the fact that carbodiazenylation (radical trapping by an aryl diazonium salt) is not a competing process. The carbodiazenylation accounts for the slightly decreased yield of 46h, as it is favoured by both the reactive diazonium salt 43c and the nucleophilicity of the intermediate alkyl and alkoxy radicals arising from the aryl radical addition to allyl acetate 45b.
Carboaminohydroxylation of vinylcarbonate proceeded with moderate diastereoslectivity, but with methylcyclohexene-1-carboxylate only one product isomer was detected. The attempted addition of phenylacetylene failed and generated the unstable benzoine derivative.7
Allylation and vinylation of aryl radicals can also be accomplished through the use of diazonium salts.24 A key element to achieve selectivity in the reaction of aryl radicals appears to be the use of water as a solvent, or at least co-solvent. Heinrich et al. achieved arylation reactions in good yields by adjusting the concentration of diazonium salt, as these act as efficient nitrogen-centred radical scavengers.24 The olefinic substrates employed were chlorides and bromides. A general reaction is described in Scheme 6.
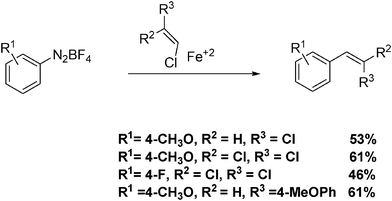 | ||
| Scheme 6 Radical reactions of diazonium salts with vinyl chlorides in water. | ||
2.6. Radical conjugate addition
The reaction of trialkylboranes with 1,4-benzoquinones to give 2-alkylhydro-quinones in quantitative yield was the first reaction of this type occurring without the assistance of a metal mediator.25a,b An ionic mechanism was originally proposed but rapidly refuted since the reaction is inhibited by radical scavengers such as galvinoxyl and iodine. Then, it was demonstrated that trialkylboranes, readily available via hydroboration, are excellent reagents for conjugate addition to vinyl ketones,26 acrolein, α-methylacrolein, α-bromoacrolein, 2-methylenecyclohexanone, and quinones (Fig. 2). Traces of oxygen present in the reaction mixture are sufficient to initiate these reactions. Their free-radical nature was demonstrated by their complete inhibition in the presence of free-radical scavengers such as galvinoxyl (5 mol%).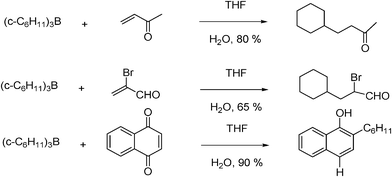 | ||
| Fig. 2 Radical conjugate additions of cyclohexyl boranes to different α,β-unsaturated compounds. | ||
The mechanism of these reactions is illustrated in Fig. 3.
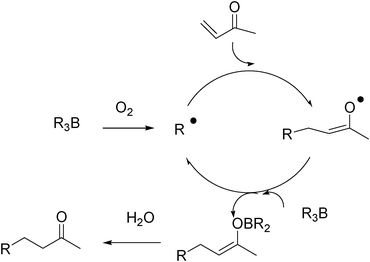 | ||
| Fig. 3 Mechanism for the radical conjugate addition of alkyl boranes to α,β-unsaturated compounds. | ||
Shea proposed a mechanism where the enolate radical resulting from the radical addition reacts with the trialkylborane to give a boron enolate27 and a new alkyl radical that can propagate the chain (Fig. 3). The formation of the intermediate boron enolate was confirmed by 1H NMR spectroscopy. The role of water present in the system is to hydrolyse the boron enolate and to prevent its degradation by undesired free-radical processes. This hydrolysis step is essential when alkynones and acrylonitrile are used as radical traps since the resulting allenes or keteneimines, respectively, react readily with radical species. Recently, Walton has shown by 11B NMR, 1H NMR, and IR spectroscopy that triethylborane does complex methyl vinyl ketone, acrolein, and 3-methylbut-3-en-2-one. They proposed that the reaction of triethylborane with these traps involves complexation of the trap by the Lewis acidic borane prior to conjugate addition (Fig. 4).28
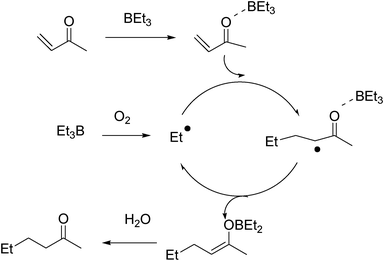 | ||
| Fig. 4 Complete proposed mechanism for the radical conjugate addition of alkyl boranes to α,β-unsaturated compounds. | ||
The reaction between trialkylboranes and enones has not found many synthetic applications.26 An exception is the preparation of prostaglandin precursors from exo-methylene cyclopentanone, generated in situ from a Mannich base. After dehydrogenation, a second conjugate addition of trioctylborane was used to introduce the ω-chain.
Synthetically, a serious drawback of the trialkylborane approach is that it requires a 1:1 trialkylborane/radical trap ratio to obtain good yields. Therefore, the method is restricted to trialkylboranes obtained by hydroboration of easily available and cheap alkenes. To overcome this limitation α-alkyl-boracyclanes have been used. According to Brown and Suzuki and co-workers, 3,3-dimethyl-borinane, prepared from BH3 and 2,4-dimethyl-1,4-pentadiene, is the most efficient reagent. With this system, selective cleavage of the boron-alkyl bond is possible for secondary and tertiary alkyl groups. This method, referred to later as the Brown–Negishi reaction,28b is not suitable for primary alkyl radicals (yield <35%) and for radical traps substituted at the β-position. With these traps, the addition of extra oxygen is necessary to run the chain reaction, and under these conditions the cleavage of the carbon–boron bond is no longer selective.
The use of ynol ethers in water is very scarce. However, the radical reaction between ethyl iodoacetate and an ynol ether has been explored successfully in water by Daoust et al., and the method is currently regarded as the first to allow the production of enol ethers in water.29
The electrophilic radical 48, formed by homolytic cleavage of activated methylenes 47 (Scheme 7, Z = EWG, electron withdrawing group) adds to the electron-rich β-carbon of ynol ether 49 leading to vinyl radical 50, whose configuration is governed by stereoelectronic factors. The use of iodides, which are efficient halogen-atom transfer agents, ensures rapid trapping of vinyl radical 50 before its interconversion. The result is an E isomer 51 obtained with high stereoselectivity. The radical initiator is Et3B in methanol (Scheme 7).
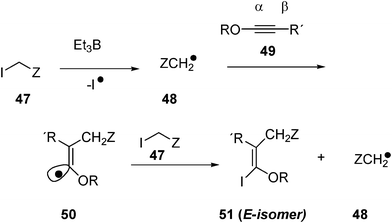 | ||
| Scheme 7 Enol ether formation in water. | ||
The reaction has to be carried out by introducing the iodoacetate and Et3B very slowly to the reaction mixture, in order to avoid the homocoupling of •CH2CO2Et radicals. The reaction proceeds with high regio- and stereoselectivity.
Different ynol ethers, 49, were used (R = Et, R' = H; R = menthyl, R′′ = H; and R = menthyl, R′′ = Me) with ethyliodoacetate (48, Z = COOEt) as the source of electrophilic radical, and the yields of substituted enol ethers 51 range from 70 to 80% in water. Iodoacetonitrile reacts modestly, while iodoacetamide adds poorly. The reaction does not occur with very bulky triple bonds. Ethyl bromoacetate, methyl bromide, and methyl iodide do not add to ynol ethers in water.
Renaud and collaborators have shown that treatment of trialkylboranes with ethenyl and ethynyloxiranes in the presence of a catalytic amount of oxygen affords the corresponding allylic or allenic alcohols. The mechanism may involve the addition of alkyl radicals to the unsaturated system leading to 1-(oxiranyl)-alkyl and 1-(oxiranyl)-alkenyl radicals followed by rapid fragmentation to give alkoxyl radicals that finally complete the chain process by reacting with the trialkylborane (Fig. 5).26
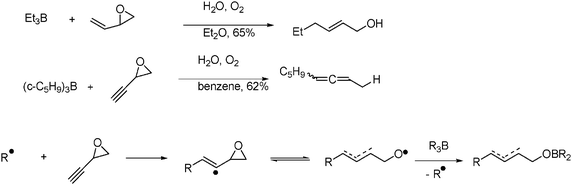
2.7. Synthesis of cycloalkane carboxylic acids in water
The aqueous carboxylation of a series of cycloalkanes has been undertaken by Pombeiro et al.30The carboxylation reactions of cycloalkanes are studied in stainless steel autoclaves by allowing the mixture to react, at low temperature and in a neutral H2O/MeCN medium, with a cycloalkane with carbon monoxide, potassium peroxodisulfate and water, either in the absence (metal-free) or in the presence (metal-promoted) of a metal promoter (eqn (11)).
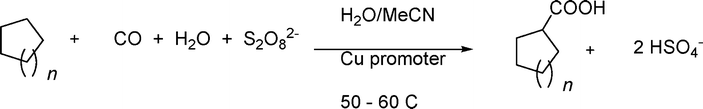 | (11) |
A crucial role in the unusual metal-free and mild transformation of an inert alkane is played by the active radical sulphate SO4− (known as an efficient single electron-transfer oxidant).
Important features of this reaction consist of the use of water as the medium, the fact that the source of the OH functionality of the carboxylic acid moiety comes from water, the possibility of working in the absence of metals, and mild temperatures (considering the high inertness of C–H bonds in cycloalkanes). Also, high efficiency and selectivity have been invoked as salient aspects of this methodology.
Among those metal compounds that exhibit a promoting effect, various derivatives of copper and potassium dichromate revealed the highest activity, leading to yields of C6H11COOH in the 70% range. The formation of cyclohexanone and cyclohexanol as by-products due to the partial oxidation of cyclohexane is also detected. The most effective promoting behaviour is exhibited by the hydrosoluble tetracopper(II) triethanolaminate derivative [O ⊂ Cu4{N(CH2CH2O)3}4(BOH)4][BF4]2.
The authors proposed a reaction mechanism as that depicted in Scheme 8.
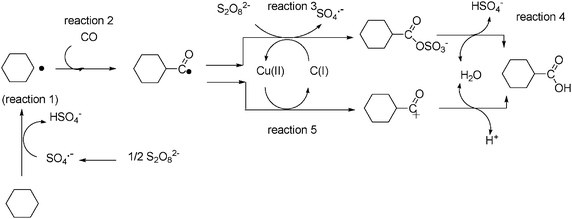 | ||
| Scheme 8 Mechanism for the synthesis of cyclohexane carboxylic acid in water. | ||
The mechanism involves the formation of a free cyclohexyl radical, which is generated by hydrogen abstraction from cyclohexane (reaction 1, Scheme 8) by the sulphate radical. The latter is derived from thermolytic and copper-promoted decomposition of K2S2O8. The involvement of cyclohexyl radical is confirmed when the reaction is carried out in the presence of the radical trap CBrCl3, which results in the full suppression of cyclohexane carboxylic acid and the complete formation of bromocyclohexane. This radical path is also inhibited by the presence of oxygen, acting as a cyclohexyl trap to afford the C6H11OO• peroxyl radical. Subsequent carbonylation of the cyclohexyl radical by carbon monoxide results in the acyl radical C6H11CO• (reaction 2, Scheme 8), that upon oxidation with further S2O82− generates the acyl sulphate C6H11C(O)OSO3− (reaction 3, Scheme 8). This is hydrolysed by water (reaction 4) generating the cyclohexane carboxylic acid. In the copper-promoted process, an alternative route (reaction 5, Scheme 8) can occur, where the tetracopper complex ([O ⊂ Cu4{N(CH2CH2O)3}4(BOH)4][BF4]2) can behave as an oxidant of the acyl radical (reaction 5, Scheme 8).
2.8. Cyclization reactions in water
Although many reagents do exist for radical generation and trapping, establishing a single prevailing mechanism is not possible. However, once a radical is generated, it can react with multiple bonds in an intramolecular fashion to yield cyclized radical intermediates. The two ends of the multiple bond constitute two possible sites of reaction. If the radical in the resulting intermediate ends up outside the ring, the attack is termed “exo”; if it ends up inside the newly formed ring, the attack is called “endo.” In many cases, exo cyclization is favoured over endo cyclization (macrocyclizations constitute the major exception to this rule). 5-Hexenyl radicals are the most synthetically useful intermediates for radical cyclizations, because cyclization is extremely rapid and endo selective. Although the exo radical is less thermodynamically stable than the endo radical, the more rapid exo cyclization is rationalized by better orbital overlap in the chair-like exo transition state (Fig. 6, vide infra).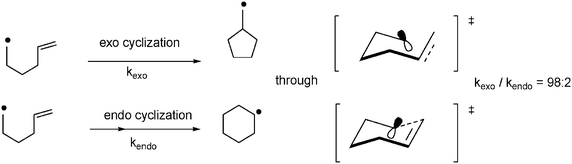 | ||
| Fig. 6 Exo versus endo cyclization of 5-hexyl radicals. | ||
Substituents that affect the stability of these transition states can have a profound effect on the site selectivity of the reaction. Carbonyl substituents at the 2-position, for instance, encourage 6-endo ring closure. Alkyl substituents at positions 2, 3, 4, or 6 enhance selectivity for 5-exo closure.
Cyclization of the homologous 6-heptenyl radical is still selective, but is much slower; as a result, competitive side reactions are an important problem when these intermediates are involved. Additionally, 1,5-shifts can yield stabilized allylic radicals at comparable rates in these systems. In 6-hexenyl radical substrates, polarization of the reactive double bond with electron-withdrawing functional groups is often necessary to achieve high yields. Stabilizing the initially formed radical with electron-withdrawing groups provides access to more stable 6-endo cyclization products preferentially.
Cyclization reactions of vinyl, aryl, and acyl radicals are also known. Under conditions of kinetic control, 5-exo cyclization takes place preferentially. However, small concentrations of a radical scavenger establish thermodynamic control and provide access to 6-endo products—not via 6-endo cyclization, but by 5-exo cyclization followed by 3-endo closure and rearrangement. Aryl radicals exhibit similar reactivity (Fig. 7).
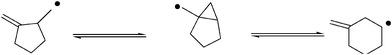 | ||
| Fig. 7 5-exo versus 6-endo cyclization. | ||
Cyclization can involve heteroatom-containing multiple bonds such as nitriles, oximes, and carbonyls. Attack at the carbon atom of the multiple bond is almost always observed. In the latter case attack is reversible; however alkoxy radicals can be trapped using a stannane trapping agent.
The use of metal hydrides (tin, silicon, and mercury hydrides) is common in radical cyclization reactions; the primary limitation of this method is the possibility of reduction of the initially formed radical by H–M. Fragmentation methods avoid this problem by incorporating the chain-transfer reagent into the substrate itself—the active chain-carrying radical is not released until after cyclization has taken place. The products of fragmentation methods retain a double bond as a result, and extra synthetic steps are usually required to incorporate the chain-carrying group. In this account we shall deal with carbon-centred radical cyclizations in water. Radical cyclizations in water involving metal-centred radicals or where other non-carbon radicals participate are not discussed herein.
Atom-transfer methods rely on the movement of an atom from the acyclic starting material to the cyclic radical to generate the product. These methods use catalytic amounts of weak reagents, preventing problems associated with the presence of strong reducing agents (such as tin hydride). Hydrogen- and halogen-transfer processes are known; the latter tend to be more synthetically useful (Fig. 8).
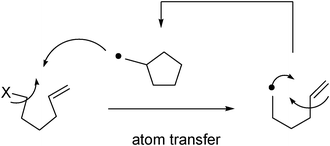 | ||
| Fig. 8 Atom transfer radical cyclization (ATRC). | ||
Oxidative and reductive cyclization methods also exist. These procedures require fairly electrophilic and nucleophilic radicals, respectively, to proceed effectively. Cyclic radicals are either oxidized or reduced and quenched with either external or internal nucleophiles or electrophiles, respectively.
The use of radicals in organic chemistry has increased substantially within the last three decades. Several methods that lead to the synthesis of carbon-centred radicals have been developed. Among the radical initiators employed, Et3B-induced radical cyclization reactions have attracted the attention of chemists. Firstly, the reactions with this initiator can be conducted at low temperatures, such as −78 °C, in the presence of trace amounts of oxygen. Secondly, various solvents including alcohol and water could be used because of the stability of Et3B in aqueous media. Among the reactions performed with this radical initiator in water, cyclizations were shown to be more efficient than in organic solvents.
Oshima et al.32 reported that the Et3B-mediated radical cyclization reactions are much more efficient in water than in benzene or hexane.31,33,34
Indeed, treatment of allyl iodoacetate 52a (eqn (12)) with Et3B in benzene or hexane does not give a lactone 53a. In contrast, in water, 52a cyclized to give 53a efficiently (67–78%). This powerful solvent effect operates also in crotyl iodoacetate and 2-pentenyliodoacetate.
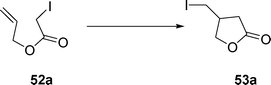 | (12) |
3-Butenyl iodoacetate 54 gave δ-lactone 55 (eqn (13)) which is generated through 6-exo cyclization in 42% yield upon treatment with Et3B in water.
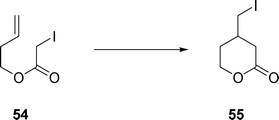 | (13) |
Treatment of α-iodoester 56 with Et3B in water provided 9-membered lactone 57 in 68% yield (eqn (14)).
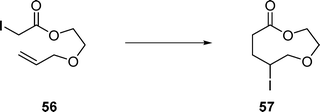 | (14) |
As a matter of fact, larger member rings are facilitated in water than in organic solvents, as is observed in Scheme 9.31
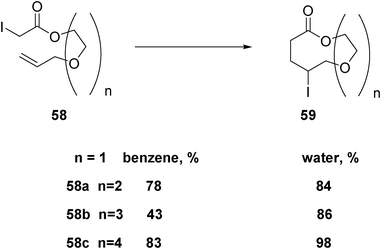 | ||
| Scheme 9 Radical formation of large-membered rings in different solvents. | ||
The authors speculate that water can effectively decrease the barrier to rotation between the major Z-rotamer Z-1 and minor E-rotamer E-1 which can cyclize (Scheme 10), whereas in benzene, the Z and E conformers of the radical do not interconvert.
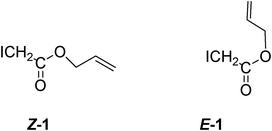 | ||
| Scheme 10 Rotamers from iodovinyl acetate. | ||
The cyclization of N-allyl iodoamides induced by Et3B proceeds smoothly in water to afford γ-lactams in good yields (Scheme 11).35
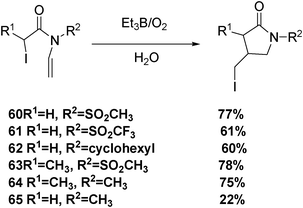 | ||
| Scheme 11 Production of pyrrolidin-2-ones in water. | ||
The low yield of the reaction from substrate 65 (Scheme 11) might be ascribed to the presence of a disfavoured conformation for cyclization. Examination of 1H NMR spectrum of 65 in D2O proved that two methyl signals (in a ratio of 42:58) appeared at δ = 2.82 and 3.02 ppm, respectively. The presence of two methyl signals is caused by restricted rotation around the C(O)–N bond attributed to the resonance form of C(O−)=N+ (Scheme 12).
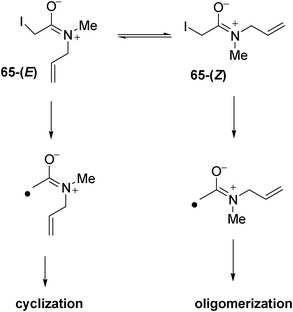 | ||
| Scheme 12 Resonance forms from N-allyl amides. | ||
Stirring a mixture of N,N-diallyl-2-iodoaetamide 66 and azo initiators at 75 °C in water yielded γ-lactam 67 (80–99% yield, eqn (15)).
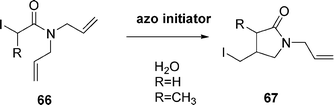 | (15) |
The best salt proved to be the N-ethylpiperidinium salt (EPHP). Dialkyl phosphonates were also examined, but they require initiation by peroxides and can lead to undesired by-products.
This development rapidly spawned several new contributions. In particular, Jang—who contributed to the initial work—showed that the sodium salt of hypophosphorous acid could reduce water-soluble organohalides in water.36,37
Jang also introduced dibutylphosphine and diphenylphosphine oxides as new reducing agents. Because they are not ionic, they are less hygroscopic than EPHP and thus could be used with water-sensitive substrates. In any case, deoxygenation of hindered substrates was possible. Comparison of the yields to those obtained via Barton's method shows that the three mediators are complementary.31
Once these two main families of P-based mediators had been introduced, rapid progress arose. Murphy reported that EPHP could trigger formation of carbon–carbon bonds either through a 6-exo-trig cyclization of an aryl radical obtained from an iodide (Murphy).38,39
Oshima introduced deuterated hypophosphorous acid potassium salts to achieve radical deuteration. Deuteration of hydrophobic substrates was possible, albeit the incorporation of deuterium was not optimal because of hydrogen atom abstraction from either the solvent or the various additives used. As water is not prone to transfer a deuterium atom, less hydrophobic substrates led to deuteration with total incorporation.
Kita built on the previous studies to report EPHP-mediated cyclization of hydrophobic substrates in water (vide infra). This breakthrough was made possible by running the reaction in the presence of a water-soluble initiator (2,2′-azobis[2-(imidazolin-2-yl)propane , VA-061) and a surfactant (cetyltrimethylammonium bromide, CTAB).40 The authors explain this outstanding result by a micellar effect generated by CTAB. The organic ammonium probably contributes to the incorporation of the hypophosphoric acid in the micelles. By trapping hypophosphorous acid with a tertiary amine, Jang introduced a surfactant-type chain carrier and reported good yields for deoxygenations of alcohols in water, without additive.36
The use of phosphorous acid as mediator in radical cyclization reactions in water can be illustrated by the synthesis of indoles from thioanilides, Scheme 13.41
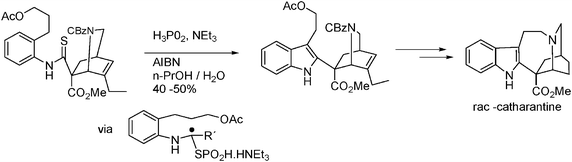 | ||
| Scheme 13 Synthesis of rac-catharantine in water. | ||
The use of 1-ethylpiperidine hypophosphite (EPHP), and the azo compound 2,2′-azobis[2-(imidazolin-2-yl)propane (VA-061) as initiator, together with a surfactant (cetyltrimethylammonium bromide, CTAB) provide the best yields of cyclized products, eqn (16).40 Several cyclizations in water involving phosphorous as radical transporters and reducing agents have been reviewed recently.31
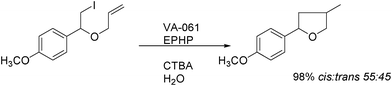 | (16) |
Upon using diethylphosphine oxide (DEPO), one can carry out sophisticated tin-free tandem radical reactions. Because DEPO is more lipophilic than hypophosphorous acid yet still water-soluble, it can facilitate the interaction between the water-soluble mediator and initiator and the lipophilic substrates without requiring a phase-transfer agent. Moreover, its pKa is 6, thus ensuring that this almost neutral excess reagent can be extracted into base during workup.42,43
Eventually, Murphy introduced a water-soluble phosphine oxide which permits higher isolated yields than the corresponding reaction using EPHP, with no additional additive. Murphy and collaborators43 have used diethylphosphine oxide (DEPO) as a radical mediator in the preparation of indolones in water, in good yields, through a sequence of aryl radical formation (R•), hydrogen atom abstraction (R–H) (Fig. 9), cyclization and rearomatization.
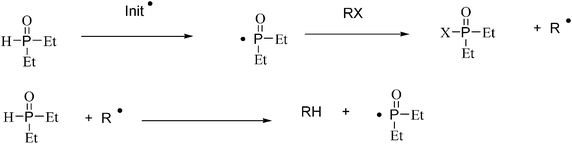
Several indolones in water could be synthesized with this methodology, and the general reaction mechanism is depicted in Fig. 10.
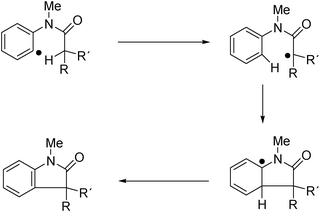 | ||
| Fig. 10 Reaction mechanism for indolone formation from aryl radicals, with ulterior rearomatization. | ||
The initiator that gave the best reaction yields is shown in Fig. 11.
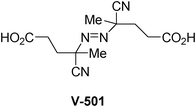 | ||
| Fig. 11 Structure of initiator V-501. | ||
Arylation of lactams in water was obtained by the use of DEPO and V-501 as the initiator in water (Fig. 12).41,44
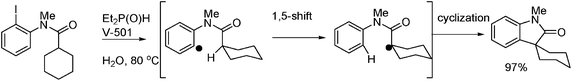
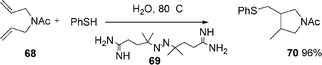
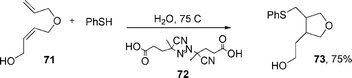
Oshima et al. also described45 that the addition of PhSH to alkenes or alkynes proceeds smoothly to give the corresponding adducts (eqn (17)–(18), see also section 3). Also, the atom transfer cyclization of diallyl-2-iodoacetamide affords lactams in excellent yields eqn (19), and the addition of 2-iodoacetamide to alkene followed by ionic cyclization gives lactone in excellent yields.
Thus, heating a mixture of N,N-diallylacetamide 68, benzenethiol, and azo initiator 69, eqn (17), in water at 60 °C, provided N-acetylpirrolidine 70 in 96% yield, eqn (17). Treatment of diallylic ether 71 with benzenethiol in the presence of the azo compound 72, eqn (18), at 75 °C, afforded tetrahydrofuran derivative 73 in 75% yield (see also section 3. for sulphur-centred radicals in water).
 | (17) |
 | (18) |
Radical cyclizations in water are successfully accomplished also by silyl radical mediators such as tris(trimethylsilyl)silane, (Me3Si)3SiH, as is depicted in Scheme 14, using 1,1′-azobis(cyclohexanecarbonitrile), ACCN, as the radical initiator.46
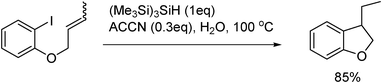 | ||
| Scheme 14 (Me3Si)3SiH-mediated radical cyclization of 1-allyloxy-2-iodobenzene in water. | ||
Postigo and collaborators,34 subjected 6-bromo-3,3,4,4,5,5,6,6-octafluoro-1-hexene (12 mM) 74 to reaction (24 h) with (Me3Si)3SiH (8 mM) and dioxygen in water (5 mL), and obtained the exo-trig cyclization product 1,1,2,2,3,3,4,4-octafluoro-5-methylcyclopentane 75 (Scheme 15) in 76% yield.
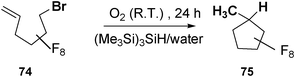 | ||
| Scheme 15 (Me3Si)3SiH-mediated radical cyclization of 6-bromo-3,3,4,4,5,5,6,6-octafluoro-1-hexene in water. | ||
Though the rate constant for cyclization in the heterogeneous water system is difficult to obtain, the cyclohexane cyclized product has not been observed in water under the reaction conditions reported. No uncyclized-reduced product is observed either.46
Analogously, cyclization of 5-bromo-1,1,2,3,3,4,4,5,5-nonafluoro-pent-1-ene (12 mM) 76 in water triggered by (Me3Si)3SiH (8 mM)/dioxygen leads to nonafluorocyclopentane, the exo-trig cyclization product 77 in 68% yield (isolated). No reduced product could be isolated from the reaction mixture. The reaction carried out in benzene-d6 does not lead to cyclization product (Scheme 16).34
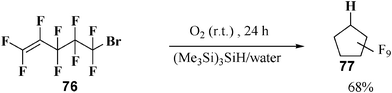 | ||
| Scheme 16 (Me3Si)3SiH-mediated radical cyclization of 5-bromo-1,1,2,3,3,4,4,5,5-nonafluoro-pent-1-ene in water. | ||
When 3,3,4,4-tetrafluoro-1,5-hexadiene (40 mM) 78 is allowed to react (24 h) in water with(Me3Si)3SiH (5 mM)/dioxygen and C2F5I (10 mM), product 79 is obtained in 61% yield, based on C2F5I (Scheme 17).34
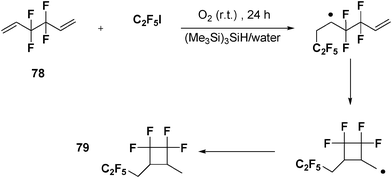 | ||
| Scheme 17 (Me3Si)3SiH-mediated radical cyclization of 3,3,4,4-tetrafluoro-1,5-hexadiene in water. | ||
Barata-Vallejo and Postigo also subjected electron-rich diallyl ether (3-(allyloxy)-prop-1-ene)11 to the radical perfluoroalkylation reaction in water with n-C6F13I (ACCN-initiation) and obtained the perfluoroalkylated tetrahydrofuran derivative in 68% yield, eqn (19), in a cis:trans ratio of ca. 80:20. Exo-cyclization of the 6-hexenyl radical is a useful probe for the elucidation of the radical mechanism of the reaction in water, as has been observed for the cyclization of a 1-allyloxy-2- iodobenzene derivative in water as well.11,34,46
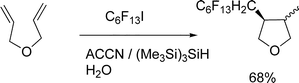 | (19) |
Newcomb, Horner, and collaborators47 have measured the intramolecular rate constant for cyclization of radical 81b in acetonitrile–water, and found a value of k = 4 × 107 s−1, eqn (20).
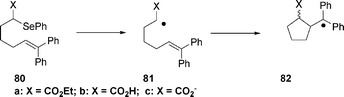 | (20) |
Radical anion 81c, formed from the photolysis of 80c in acetonitrile–water solution eqn (20), gave distinctly different results. The rate constant for cyclization of 81c at 20 °C was k = 3.5 × 106 s −1 at pH 7.4, an order of magnitude smaller than the rate constant for cyclization of 81b. The pronounced effect of the carboxylate group in 81c is noteworthy because the identity of the X group in other radicals 81 has little effect on the rates of cyclization; for radicals 81 with X = H, CH3, OCH3, CO2Et, CONEt2, and CO2H, the rate constants for cyclization at 22 °C are in the range k = 2–5 × 107 s−1 . A temperature-dependent kinetic study performed by the authors40 showed that the reduction in the rate constant for cyclization of radical anion 81c is due to an enthalpy effect in the transition state that slows the reaction, even though the entropy of activation for the reaction is more favourable than those for cyclizations of analogues with other X groups. Specifically, the Ea (activation energy) for cyclization of radical anion 81c, eqn (20), at pH 7.4 of 6.5 kcal mol−1 is ca. 3 kcal mol−1 greater than that found for any other 81 radical thus far studied. The authors47 speculate that charge delocalization in radical anion 81c and subsequent localization upon cyclization to 82c is an important enthalpy feature.
The significant difference in rate constants for cyclizations of radical 81b and radical anion 81c detected by a kinetic titration study47 that demonstrated the robust nature of the approach at various pH. Reactions were conducted in buffered acetonitrile–water solutions, and the observed rate constants were fitted to eqn (21), where kA and kB are rate constants for the cyclization of the acid (81b) and basic (81c) forms, respectively, and Ka is the acidity constant for 81b.
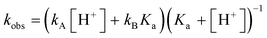 | (21) |
Regression analysis gave kA = (3.43 ± 0.08) × 107 s −1 , kB = (3.4 ± 1.0) × 106 s −1 , and Ka = (2.3 ± 0.4) × 10−5. The apparent pKa of α-carboxylic acid radical 81b in water is 4.6 . The α-radicals from small alkanoic acids have pKa values similar to those of the parent acids. Access to α- and β-carboxylate radicals could be important for studies of rearrangements catalysed by coenzyme B12-dependent enzymes, as suggested by the authors47 because it can produce model radical anions that closely resemble the reactive species in nature. The large effect of the carboxylate group in the kinetic parameters for the cyclization of 81b, a reaction that might be expected to have low sensitivity to charge effects, suggests that neutral ester and carboxylic acid radicals might be poor models for radical anions. The authors47 speculated that the negative charge in the radical anions is a critically important feature in rearrangements catalysed by coenzyme B12-dependent enzymes, one that permits heterolytic fragmentation reaction pathways that cannot be accessed from neutral radicals.
The radical–polar crossover reaction48 in Scheme 18 affords a means of performing radical-based carbon–carbon bond formation. Thus, tetrathiafulvalene (TTF, Scheme 18) donates an electron to the diazonium group, which upon loss of dinitrogen affords an aryl radical which cyclizes, and the resulting radical couples with TTF+ to form a sulfonium salt, which undergoes solvolysis to afford alcohol 84 or an amide 83 affording a polar termination to the radical reaction (Scheme 18).
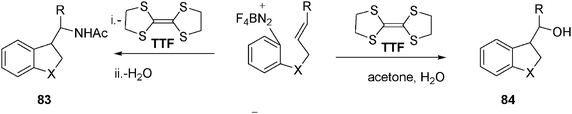 | ||
| Scheme 18 Reaction of diazonium salts with tetrathiafulvalene (TTF) in water. | ||
More complex amines 85 and 86 can be converted into the corresponding diazonium tetrafluoroborates through the use of a water-soluble tetrathiafulvalene-derivative, eqn (22), which then successfully undergo radical cyclization, radical–polar crossover and intramolecular termination in 1:1 acetone:water, demonstrating the scope of the reaction.49
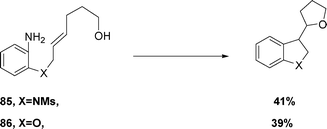 | (22) |
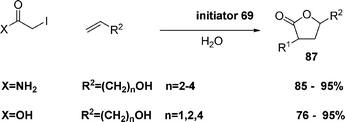 | (23) |
The authors50 suggested a mechanism such as that proposed in Scheme 19.
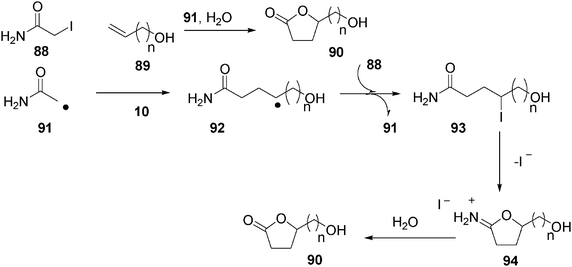 | ||
| Scheme 19 Mechanism proposed for the formation of lactones from iodoacetamides and alkenols in water | ||
Radical 91 (derived from thermal treatment of iodide 88 in the presence of azo compound 69, see the structure in eqn (17)), adds readily to the alkenyl terminal carbon of alkenol 89 to render radical 92. The iodine atom transfer between radical 92 and iodide 88 affords a hydroxy-4-iodoamide 93, and regenerates radical 91. Compound 93 cyclizes to γ-lactone 90via94 under the reaction conditions due to the well-known ionic lactonization of 4-iodoalkanamide (Scheme 19).
Interestingly enough, the same authors argue that the solvent effect of water is exerted in the initiation step, activating the carbon–iodine bond in 88.
Landais and co-workers reported on the radical addition-5-exo-trig cyclization on ketoximes, Scheme 20.50
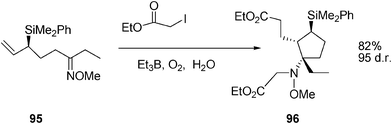 | ||
| Scheme 20 Radical cyclization of ketoximes in water. | ||
For instance, when the radical reaction was carried out on 95, and water was used as a solvent, this led to the unique formation of diester 96. Similar results were obtained when additives such as AcOH (1–5 equiv.) were employed. Strikingly, the reaction can even be conducted in 1 N HCl or in conc. H2SO4 and still leads to the exclusive formation of 96 (albeit at lower chemical efficiency). Electrophiles such as (Boc)2O and trifluoroacetic anhydride were also tested to trap the putative amidoborane (Scheme 21), but 96 was invariably produced as a unique product. These experiments clearly rule out the formation of the C–N bond and the incorporation of the second ester fragment through an ionic pathway.50 The reaction mechanism is depicted in Scheme 21.
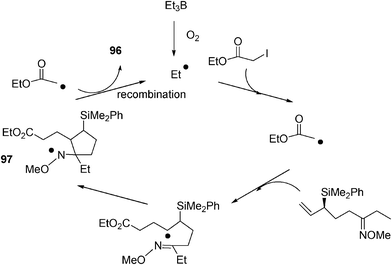 | ||
| Scheme 21 Mechanism for the formation of product 96 in water. | ||
Precedents from the literature effectively reveal that alkoxyaminyl radicals (such as 97) are persistent due to steric shielding.51,52 In the case depicted in Scheme 21, intermediate radical 97 may fit into this category and therefore possess a long enough lifetime to recombine with an excess of radical precursor (EtOCOCH2) arising from the iodoester. Several experiments were designed to reduce the alkoxyaminyl radical intermediate 97 under free-radical conditions.
For instance, addition of ethyl xanthate in isopropanol as both a solvent and a reducing agent, according to Zard's procedure,53 led to 96 in low yield (8%) along with recovered starting material (78%). No trace of the desired reduced product was observed. The use of Roberts's polarity-reversal catalysis54–57 (Ph3SiH, HSCH2CO2Et (cat.), BrCH2CO2Et) led in turn to a complex mixture of products in which the desired product was absent. Finally, substitution of Et3B for indium in a MeOH–H2O mixture led to recovered allylsilane 95 and ethyl acetate resulting from complete reduction of ethyliodoacetate. In parallel, the authors envisaged being able to trap the alkoxyaminyl radical (97) in an intramolecular fashion through the introduction of an olefinic appendage. Recent studies effectively showed that a 5-exo-trig cyclization onto a ketoxime, followed by a second 5-exo-trig addition of an alkoxyaminyl radical onto an unsaturated Michael acceptor, was feasible, leading to a good yield of the desired bicyclic system in water.
The one-pot radical addition of the iodomalonate 98 to 1-octene in water followed by successive cyclization and azidation afforded the tertiary azide 99 in 72% yield as a 4:1 mixture of diastereomers, eqn (24).
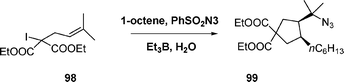 | (24) |
The radical cyclic addition of cyclic 1,3-dicarbonyl compounds to the appropriate olefins provides a versatile method for the synthesis of fused dihydrofuran derivatives.58 When electron-poor alkenes were employed as the substrates in aqueous mixtures, the radical reaction pathway resulted in the regioselective generation of product 100 with the carbon of 1,3-dicarbonyl compounds added at the α-position of electron-poor alkenes (path a, Fig. 13). Wang and co-workers reported a Mn(OAc)3-mediated reversed regioselective radical cyclic addition (path b, Fig. 13). However, according to the plausible reaction pathway, only 1-(pyridin-2-yl)enones were suitable substrates.59
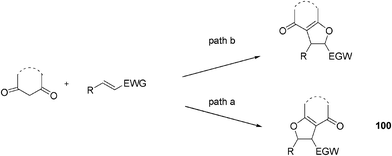 | ||
| Fig. 13 Radical synthesis of dihydrofurans from 1,3-dicarbonyl compounds. | ||
Also, oxime ethers can undergo tandem radical cyclization in water.16 Treatment of oxime ether 101 with Et3B and isopropyl iodide in water at 80 °C affords the cyclized product 102 in 63% yield via two carbon–carbon bond-forming steps, eqn (25).
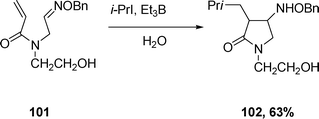 | (25) |
More recently, Fan and coworkers60 reported on radical cyclization using a Michael adduct, and found that the best conditions entailed the use of PhIO, Bu4NI in water, as shown in Scheme 22.
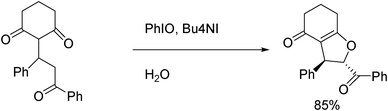 | ||
| Scheme 22 Radical synthesis of fused dihydrofurans in water. | ||
The scope of this reaction was then investigated under optimized conditions (those from Scheme 22), and the results were very promising. The Michael adducts of chalcone with 5,5-dimethylcyclohexane-1,3-dione (dimedone), 6-methyl-3H-pyran- 2,4-dione, chroman-2,4-dione (4-hydroxycoumarin), and 6-fluorochroman-2,4-dione (4-hydroxy-6-fluorocoumarin) were effective substrates, and their reactions gave rise to the corresponding fused dihydrofurans in good to excellent yields (up to 90% yield).60 This is an efficient method developed for the construction of functionalized fused dihydrofurans via an aqueous PhIO/Bu4NI-mediated stereoselective oxidative cyclization of Michael adducts of cyclic 1,3-dicarbonyl compounds with chalcones. The potential of this reaction system can be evaluated by its simple procedure, mild conditions, and adaptability to a wide variety of substrates.
3. Sulphur-centred radicals in water
3.1. Thioesterification with aldehydes
Kita et al.61 reported on an effective intermolecular radical reaction for carbon–sulphur bond formation in a micellar system using the combination of a water-soluble radical initiator and surfactant in water (Scheme 23).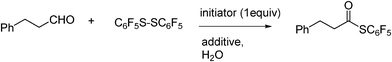
The initiator that gave the best reaction yields is VA-044 (1,2-bis(2-(4,5-dihydro-1H-imidazol-2-yl)propan-2-yl)diazene hydrochloride), and the additive used is the cationic surfactant CTAB. When galvinoxyl free radical was used as a radical scavenger, no thioesterification occurred. Hence, this reaction proceeds via a radical mechanism. Water was also shown to be the best solvent for the reaction.
Various disulphides were used in the above reaction, among which, pentafluorodiphenyl sulphide gave the corresponding thioesters in best yields. Among aldehydes, aliphatic aldehydes and aromatic aldehydes with electron releasing groups afforded the best thioester yields.
A plausible reaction mechanism is depicted in Scheme 24.
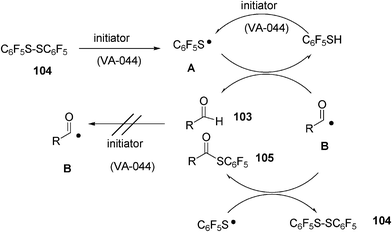
The disulphide 104 dissociates upon reaction with the initiator to yield thiyl radicals A. Secondly, the hydrogen from the aldehyde 103 is trapped by the thiyl radical A and the acyl radical B is formed. The acyl radical reacts with the disulphide 104 or thiyl radical A, and the thioester 105 is formed (Scheme 24).
The authors also achieved the direct amidation of aldehydes in water using a one-pot synthetic methodology. Several aldehydes such as p-methoxybenzaldehyde, 2,4,6-trimethylbenzaldehyde, 2,4,6-trimethoxybenzaldehyde, 3,4-dimethoxybenzaldehyde were used in reactions.62
3.2. Hydrogen abstraction and cis–trans double bond isomerization by thiyl radicals in water
Zhao et al.63 have derived rate constants for H-abstraction by cysteine thiyl radicals at pH 10.5 from anionic glycine (3.2 × 105 M−1 s−1) and alanine (7,7 × 105 M−1 s−1). The deprotonated amino group ensures optimum captodative stabilization of the αC• radical. However, deprotonated aliphatic amines are physiologically unrealistic.Schöneik et al.64a reported an H atom abstraction reaction from peptides by thiyl radicals, directly relevant for aminoacids within proteins. The substrates in their work cover a broad range of αC–H bond energies. For example, in N-formyl-Asp-NH2, the calculated BDE(αC–H) = 332 kJ mol−1, and in N-acetyl-Pro-NH2, BDE(αC–H) = 369 kJ mol−1 (trans-Pro) and 358 kJ mol−1 (cis-Pro).
Hydrogen abstraction by thiyl radicals in water have been documented in several instances, especially from silanes in polarity reversal catalysis.33,34,4,54,56,57
Cis–trans double bond isomerization by thiyl radicals has been established by Chatgilialoglu and collaborators both in solution and microheterogeneous media.64b–e
3.3. Synthesis of (α-fluoro)vinyl sulphides by thiodesulfonylation of vinyl sulfones
The removal of the sulfonyl group from vinylic carbon is usually carried out by reductive methods or addition–elimination processes where the sulfonyl group is replaced by a tributylstannyl substituent, which is then replaced by H.Wnuk et al.65 reported the stereoselective radical-mediated thiodesulfonylation of vinyl and (α-fluoro)vinyl sulfones in water. Such thiodesulfonylation provides a flexible alternative to the hydrothiolation of alkynes with thiols under radical or metal catalysis conditions. It also offers a convenient preparation of (α-fluoro)vinyl sulphides. This methodology can be envisaged as a reductive deoxygenation of sulfones to the corresponding sulphides.
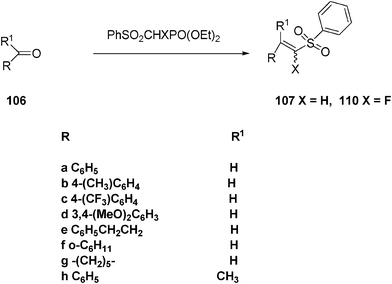
Treatment of the sulfonyl-stabilized enolates generated from diethyl (phenylsulfonyl)methylphosphonate with aliphatic and aromatic aldehydes and ketones 106a–g gave the corresponding E-vinyl sulfones 107a–f and vinyl sulfone 107g (72–95%, Scheme 25). Analogous treatment of 106a–h with diethyl fluoro(phenylsulfonyl)methylphosphonate produced (α-fluoro)vinyl sulfones 108a–h (Scheme 27).
 | ||
| Scheme 25 Synthesis of E-vinyl and (E/Z)-(α-fluoro)vinyl sulfones. | ||
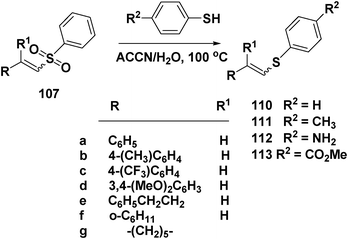
Treatment of E-107a–c with benzenethiol (R2 = H) afforded 110a–c in 95%, 54% and 85% yields respectively, when the reaction was carried out in water, and initiated by ACCN (1,1′-azo-bis-(carbonitrilecyclohexane)), Scheme 26.
 | ||
| Scheme 26 Thiodesulfonylation of vinyl sulfones. Synthesis of vinyl sulphides. | ||
Treatment of E-107a with 4-methylbenzenethiol or 4-aminobenzenethiol in H2O/ACCN, produced the corresponding vinyl sulphides 111a (61%) and 112a (55%). Analogously, E-107c was converted to 111c and 112c in 58 and 55% yields, respectively (Scheme 26).
Radical-mediated thiodesulfonylation of the vinylsulfones 107 occurred with retention of the E stereochemistry. In order to study the stereochemical outcome of the thiodesulfonylation reactions, Z-vinyl sulfones 107a was prepared by anti-Markovnikov addition of PhSH/NaOH to phenylacetylene followed by the oxidation of the resulting (Z)-2-phenyl-1-phenylthioethene. Treatment of Z-107a with PhSH in aqueous medium produced sulphide 110a in very good yields with inversion of stereochemistry (E/Z, 95:5). Thus the vinyl sulphides are formed predominantly with E-stereochemistry independently of the stereochemistry of the starting vinyl sulfones.
Thiodesulfonylation appears to be fairly general since sulfones with alkyl (methyl), electron withdrawing (CF3) and electron donating substituents (CH3O) on the phenyl ring attached to the double bond also produced (α-fluoro)vinyl sulphides (Scheme 27).
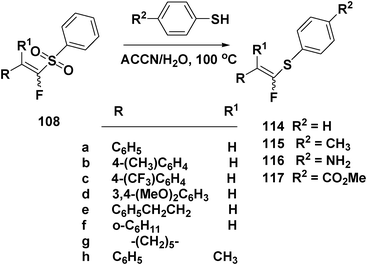 | ||
| Scheme 27 Thiodesulfonylation of (E/Z)-(α-fluoro)vinyl sulphides. | ||
Radical thiodesulfonylation permitted the synthesis of the sparsely developed (α-fluoro)vinyl sulphides 114–117 in high yields (Schemes 27, 28).
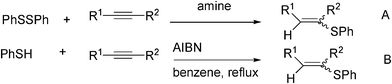 | ||
| Scheme 28 Radical synthesis of 1-akenylsulphides. | ||
Reaction of 108h (E/Z, 57:43) with benzenethiol also afforded tetrasubstituted (α-fluoro)vinyl sulphide 114h in 58% yield. Reactions of 108b–d with benzenethiol produced 114b–d in 82%, 60% and 73% yields, respectively, when the reactions are carried out in water, and initiated by ACCN. Thiodesulfonylation occurred with other aromatic thiols as well. In Scheme 27, a summary of the scope of this reaction is presented, with 4-methylbenzenethiol, and 4-aminobenzenethiol. It is noteworthy that hydrothiolation of alkynes is inapplicable for the synthesis of (α-fluoro)vinyl sulphides since the 1-fluoralkynes are unstable and virtually unknown. Desulfonylation occurred probably via β-elimination of the sulfonyl radical from the radical intermediates formed after addition of PhS• to vinyl sulfones (presumably via a radical addition–elimination mechanism).
3.4. Radical hydrothiolation of alkynes with diphenyldisulphide and tripropylamine
It is well known that the benzenethiyl radical formed from thiophenol in the presence of AIBN undergoes an addition reaction with alkynes to give vinyl sulphides.66 In this reaction, the use of ill-smelling thiophenol and a hazardous radical initiator such as AIBN was required.Although many reactions of alkynes with disulphides have been developed, disulfidation products are generally obtained.67 However, reductive radical reaction of alkynes with disulphide to give 1-alkenyl sulphides has not been explored.
Ishibashi and collaborators68 have developed a method for the formation of benzenethiyl radical from diphenyl disulphide with tripropylamine via a single electron transfer (SET) reaction in water. Inexpensive and environmentally friendly reagents were employed, and the experimental procedure is very simple and safe, according to Scheme 29.
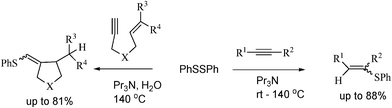 | ||
| Scheme 29 Benzenethiyl radicals from diphenyldisulphide in water. | ||
A plausible mechanism for the reaction is shown in Scheme 30. The reaction may be initiated by single electron transfer (SET) process of tripropylamine to disulphide 118 to generate an anion radical 122 and a cation radical 123. The S–S bond cleavage of anion radical 122 generates benzenethiyl radical 124 and thiolate anion 125. An attack of thiyl radical 124 on the alkyne 119a gives the vinyl radical 127, which then abstracts a hydrogen atom from benzenethiol 126 to give vinyl sulphide 120a together with the benezenthiyl radical 124. It should be noted that the formation of benzenethiol 126 might be a result of the removal of a proton of cation radical 123 (proton removal of 123 gives radical 128). Therefore, a single electron transfer reaction between tripropylamine and diphenyl disulphide 120 to form cation radical 123 is important to give vinyl sulphides 120a. Partial formation of compound 121a might be a result of an attack of vinyl radical 127 on diphenyl disulphide 118 (Scheme 30).
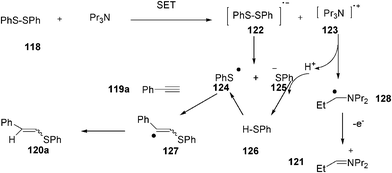 | ||
| Scheme 30 Mechanism for the radical synthesis of alkenylsulphides in water. | ||
Sulphur-based radicals, generated from the RSH-type precursor (R = alkyl, acyl) with AIBN added smoothly to allyl glycine.69 Optimal results were obtained when both the unsaturated amino acids and RSH dissolved completely in the reaction medium (dioxane–water and methanol–water where found to be superior solvents).69,70 Radical additions of thiophenol to carbon–carbon multiple bonds and radical cyclization of N-allyl-2-iodoalkanamide in aqueous media proceeded smoothly to N-acetylpyrrolidine derivative in 96% yield. Under similar experimental conditions, atom transfer radical cyclization of 2-iodo amides in water gave corresponding lactams in 96% and 85% yield (see also (eqn 17 and 18) in section 2.8.2).69
4. Concluding remarks
Radical chemistry lies at the heart of many organic synthetic transformations used to achieve fine chemicals, the synthesis of natural products and pharmaceuticals. Such a role confers radical chemistry the risk of contributing negatively to environmental pollution. However, the design and utilization of solvents that have a reduced impact on the environment and serve as alternatives to the currently used volatile organic solvents, chlorinated solvents, and solvents that damage the natural environment are considered landmarks for advancing a particular synthetic protocol. Thus the number of research articles on radical reactions in water has awakened the need for guiding chemists toward further research and discoveries.Throughout this review, advantages in the regio- and stereoselectivity of the reactions employing carbon and sulphur-centred radicals have been highlighted when water and other aqueous media are used. Interestingly, examples are provided where radical transformations in organic media utilising carbon- and sulphur-centred radicals do not lead to products in acceptable yields, whereas in water the reactions progress to the desired products in optimal yields. Of note is the finding of the enhanced efficiency of radical chain initiation in water as opposed to in organic solvents, the former being accomplished with a wider array of initiators. It is likely that, in the chain reaction, both the initiation and propagation steps benefit in water or aqueous media, either due to the volume of water, or the hydrogen-bonding ability. Undoubtedly, more research in this area is necessary.
Radical atom transfers reactions (such as those studied in section 2.3) are classical examples of atom efficiency in organic synthesis devoted to waste minimization. For radical rearrangement reactions (intramolecular cyclization reactions, section 2.8.1), a single reactant leads to a single product with nearly the same mass and an almost identical structure. Although cyclization reactions can be categorized as rearrangements, they generally show a decrease in (rotational) entropy, as bond rotation or the number of conformations in the cyclic product is not as great as that for the open-chain starting materials. All these factors support the good prospects for radical chemistry in water as have been reported in this account.
References
- (a) P. G. Jessop, Green Chem., 2011, 13, 1391 RSC; (b) H. Fischer and L. Random, Angew. Chem., Int. Ed., 2001, 40, 1340 CrossRef CAS; (c) G. Litwinienko and K. U. Ingold, Acc. Chem. Res., 2007, 40, 222 CrossRef CAS.
- K. U. lngold, Pure Appl. Chem., 1997, 69, 241 CrossRef.
- G. Litwinienko, G. A. DiLabio, P. Mulder, H.-G. Korth and K. U. Ingold, J. Phys. Chem. A, 2009, 113, 6275 CrossRef CAS.
- M. F. Nielsen and K. U. Ingold, J. Am. Chem. Soc., 2006, 128, 1172 CrossRef CAS.
- D. W. Snelgrove, J. Lusztyk, J. T. Banks, P. Mulder and K. U. Ingold, J. Am. Chem. Soc., 2001, 123, 469 CrossRef CAS.
- D. V. Avila and K. U. Ingold, J. Am. Chem. Soc., 1995, 117, 2929 CrossRef CAS.
- S. J. Garden, D. V. Avila, A. L. J. Beckwith, V. W. Bowry, K. U. Ingold and J. Lusztyk, J. Org. Chem., 1996, 61, 805 CrossRef CAS.
- C. Galli, Chem. Rev., 1988, 88, 765 CrossRef CAS.
- L. Zhang, W. R. Dolbier Jr., B. Sheeller and K. U. Ingold, J. Am. Chem. Soc., 2002, 124, 6362 CrossRef CAS.
- X. X. Rong, H.-Q. Pan and W. R. Dolbier, Jr., J. Am. Chem. Soc., 1994, 116, 4521 CrossRef CAS.
- S. Barata-Vallejo and A. Postigo, J. Org. Chem., 2010, 75, 6141 CrossRef CAS.
- (a) J. C. Scaiano and L. C. Stewart, J. Am. Chem. Soc., 1983, 105, 3609 CrossRef CAS; (b) L. J. Johnston, J. C. Scaiano and K. U. Ingold, J. Am. Chem. Soc., 1984, 106, 4877 CrossRef CAS.
- B. Delest, A. B. Shtarev and W. R. Dolbier, Jr., Tetrahedron, 1998, 54, 9273 CrossRef CAS.
- J. Kondo, H. Shinokubo and K. Oshima, Angew. Chem., Int. Ed., 2003, 42, 825 CrossRef CAS.
- H. Yorimitsu, H. Shinokubo, S. Matsubara and K. Oshima, J. Org. Chem., 2001, 66, 7776 CrossRef CAS.
- (a) H. Miyabe, M. Ueda and T. Naito, J. Org. Chem., 2000, 65, 5043 CrossRef CAS; (b) H. Miyabe, K. Fujii, T. Goto and T. Naito, Org. Lett., 2000, 2, 4071 CrossRef CAS.
- S. B. McNabb, M. Ueda and T. Naito, Org. Lett., 2004, 6, 1911 CrossRef CAS.
- H. Miyabe, Y. Yamaoka and Y. Takemoto, J. Org. Chem., 2005, 70, 3324 CrossRef CAS.
- M. S. Kharasch and F. R. Mayo, J. Am. Chem. Soc., 1933, 55, 2468 CrossRef CAS.
- J. M. Joung, J. H. Ahn, D. W. Lee and N.M. Yoon, J. Org. Chem., 1998, 63, 2755 CrossRef.
- P. Panchaud and P. A. Renaud, J. Org. Chem., 2004, 69, 3205 CrossRef CAS.
- Z. Tu, C. Lin, Y. Jang, Y-J. Yang, S. Ko, H. Fang, J-T. Liu and C-F. Yao, Tetrahedron Lett., 2006, 47, 6133 CrossRef CAS.
- H. R. Heinrich and A. Wetzel, Org. Lett., 2007, 9, 3833 CrossRef.
- M. Heinrich, O. Blank, D. Ullrich and M. Kirschstein, J. Org. Chem., 2007, 72, 9609 CrossRef CAS.
- (a) L. W. Bieber, P. J. Rolim Neto and R. M. Generino, Tetrahedron Lett., 1999, 40, 4473 CrossRef CAS; (b) G. S. C. Srikanth and S. L. Castle, Tetrahedron, 2005, 61, 10377 CrossRef CAS.
- C. Olivier and P. Renaud, Chem. Rev., 2001, 101, 3415 CrossRef.
- Jie Bai, L. D. Burke and K. J. Shea, J. Am. Chem. Soc., 2007, 129, 4981 CrossRef CAS.
- (a) E. Kumli, F. Montermini and P. Renaud, Org. Lett., 2006, 8, 5861 CrossRef CAS; (b) A. Suzuki, A. Arase, H. Matsumoto, M. Itoh and H.C. Brown, J. Am. Chem. Soc., 1967, 89, 5708 CrossRef CAS; (c) J. C. Walton, A. J. McCarrol, Q. Chen, B. Carboni and R. Nziengui, J. Am. Chem. Soc., 2000, 122, 5455 CrossRef CAS.
- P. Lemoine and B. Daoust, Tetrahedron Lett., 2008, 49, 6175 CrossRef CAS.
- M. V. Kirillova, A. M. Kirillov and A. J. L. Pombeiro, Adv. Synth. Catal., 2009, 351, 2936 CrossRef CAS.
- (a) V. T. Perchyonok and I. N. Lykakis, Curr. Org. Chem., 2009, 13, 573 CrossRef CAS; (b) A. Postigo, in Organic Radical Reactions in Water and Alternative Media, Nova Science Publications, 2011, Chapter III Search PubMed; (c) A. Postigo and N. Sbarbati Nudelman, Coord. Chem. Rev., 2011 Search PubMed, in revision.
- H. Yorimitzu, T. Nakamura, H. Shinokubo and K. Oshima, J. Org. Chem., 1998, 63, 8604 CrossRef.
- A. Postigo, Curr. Org. Chem., 2009, 13, 1683 CrossRef CAS.
- S. Barata-Vallejo, N. Sbarbati Nudelman and A. Postigo, Curr. Org. Chem., 2011, 15, 1826 CAS.
- K. Wakabayashi, H. Yorimitzu, H. Shinokubo and K. Oshima, Bull. Chem. Soc. Jpn., 2000, 73, 2377 CrossRef CAS.
- D. O. Jang, Tetrahedron Lett., 1996, 37, 5367 CrossRef CAS.
- D. O. Jang, D. H. Cho and J. Kim, Synth. Commun., 1998, 28, 3559 CrossRef CAS.
- C. Gonzalez Martin, J. A. Murphy and C. R. Smith, Tetrahedron Lett., 2000, 41, 1833 CrossRef.
- J. A. Murphy, R. Tripoli, T. A. Khan and U. W. Mali, Org. Lett., 2005, 7, 3287 CrossRef CAS.
- Y. Kita, H. Nambu, N. G. Ramesh, G. Anilkumar and M. Matsugi, Org. Lett., 2000, 3, 1157 CrossRef.
- D. Leca, L. Fensterbank, E. Lacote and M. Malacria, Chem. Soc. Rev., 2005, 34, 858 RSC.
- D. H. Cho and D. O. Jang, Tetrahedron Lett., 2005, 46, 1799 CrossRef CAS.
- T. A. Khan, R. Tripoli, J. L. Crawford, C. G. Martin and J. A. Murphy, Org. Lett., 2003, 5, 2971 CrossRef CAS.
- H. Yorimitsi, T. Nakamura, H. Shinokubo, K. Oshima, K. Omoto and H. Fujimoto, J. Am. Chem. Soc., 2000, 122, 11041 CrossRef.
- H. Yorimitsu, K. Wakabayashi, H. Shinokubo and K. Oshima, Bull. Chem. Soc. Jpn., 2001, 74, 1963 CrossRef CAS.
- A. Postigo, S. Kopsov, C. Ferreri and C. Chatgilialoglu, Org. Lett., 2007, 9, 5159 CrossRef CAS.
- N. Miranda, P. Daublain, J. H. Horner and M. Newcomb, J. Am. Chem. Soc., 2003, 125, 5260 CrossRef CAS.
- C. Lampard, J. A. Murphy and N. Lewis, J. Chem. Soc., Chem. Commun., 1993, 295 RSC.
- B. Patro, M. C. Merrett, S. D. Markin, J. A. Murphy and K. E. B. Parkes, Tetrahedron Lett., 2000, 41, 421 CrossRef CAS.
- E. Godineau, K. Schenk and Y. Landais, J. Org. Chem., 2008, 73, 6983 CrossRef CAS.
- A. F. Bella, A. M. Z. Slawin and J. C. Walton, J. Org. Chem., 2004, 69, 5926 CrossRef CAS.
- H. Teichert, K. Jantos, K. Harms and A. Studer, Org. Lett., 2004, 6, 3477 CrossRef.
- L. Boiteau, J. Boivin, A. Liard, B. Quiclet-Sire and S. Z. Zard, Angew. Chem., Int. Ed., 1998, 37, 1128 CrossRef CAS.
- B. P. Roberts, Chem. Soc. Rev., 1999, 28, 25 RSC.
- A. Postigo and N. Sbarbati Nudelman, J. Phys. Org. Chem., 2010, 23, 910 CrossRef CAS.
- A. Postigo, C. Ferreri, M. L. Navachia and C. Chatgilialoglu, Synlett, 2005, 2854 CAS.
- A. Postigo, S. Kopsov, C. Ferreri and C. Chatgilialoglu, Organometallics, 2009, 28, 3282 CrossRef CAS.
- M. Ceylan and E. Fíındíık, Synth. Commun., 2008, 38, 1070 CrossRef CAS.
- G.-W. Wang, Y.-W. Dong, P. Wu, T.-T. Yuan and Y.-B. Shen, J. Org. Chem., 2008, 73, 7088 CrossRef CAS.
- Y. Ye, L. Wang and R. Fan, J. Am. Chem. Soc., 2010, 75, 1760 CAS.
- H. Nambu, K. Hata, M. Matsugi and Y. Kita, Chem.–Eur. J., 2005, 11, 719 CrossRef CAS.
- H. Nambu, K. Hata, M. Matsugi and Y. Kita, Chem. Commun., 2002, 1082 RSC.
- R. Zhao, J. Lind, G. Merényi and T. E. Eriksen, J. Am. Chem. Soc., 1994, 116, 12010 CrossRef CAS.
- (a) T. Nauser and C. Schöneik, J. Am. Chem. Soc., 2003, 125, 2042 CrossRef CAS; (b) C. Chatgilialoglu and C. Ferreri, Acc. Chem. Res., 2005, 38, 441 CrossRef CAS; (c) C. Chatgilialoglu and A. Altieri; H. Fischer, J. Am. Chem. Soc., 2002, 124, 12816 CrossRef CAS; (d) C. Ferreri, A. Samadi, F. Sassatelli, L. Landi and C. Chatgilialoglu, J. Am. Chem. Soc., 2004, 126, 1063 CrossRef CAS; (e) I. N. Lykakis, C. Ferreri and C. Chatgilialoglu, Angew. Chem., Int. Ed., 2007, 46, 1914 CrossRef CAS.
- P. R. Sacasa, J. Zayas and S. F. Wnuk, Tetrahedron Lett., 2009, 50, 5424 CrossRef CAS.
- L. Benati, P. C. Montevecchi and P. Spagnolo, J. Chem. Soc., Perkin Trans. 1, 1991, 2103 RSC.
- V. P. Ananikov, M. A. Kabeshov, I. P. Beletskaya, V. N. Khrustalev and M. Y. Antipin, Organometallics, 2005, 24, 1275 CrossRef CAS.
- T. Taniguchi, T. Fujii, A. Idota and H. Ishibashi, Org. Lett., 2009, 11, 3298 CrossRef CAS.
- H. Yorimitsu, K. Wakabayashi, H. Shinokubo and K. Oshima, Tetrahedron Lett., 1999, 40, 519 CrossRef CAS.
- S. Mikami, K. Fujita, T. Nakamura, H. Yorimitsu, H. Shinokubo, S. Matsubara and K. Oshima, Org. Lett., 2001, 3, 1853 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |