Chalcogen-based aerogels as a multifunctional platform for remediation of radioactive iodine
Brian J.
Riley
*a,
Jaehun
Chun
a,
Joseph V.
Ryan
a,
Josef
Matyáš
a,
Xiaohong S.
Li
a,
Dean W.
Matson
a,
Shanmugavelayutham K.
Sundaram
b,
Denis M.
Strachan
a and
John D.
Vienna
a
aNon-Oxide Materials Synthesis Laboratory, Pacific Northwest National Laboratory, Richland, WA 99352, USA. E-mail: brian.riley@pnnl.gov; Fax: +1 (509)372-5997; Tel: +1 (509)372-4651
bCeramic Engineering Department, Alfred University, Alfred, NY 14802, USA
First published on 27th October 2011
Abstract
Aerogels employing chalcogen-based (i.e., S, Se, and/or Te) structural units and interlinking metals are termed chalcogels and have many emerging applications. Here, chalcogels are discussed in the context of nuclear fuel reprocessing and radioactive waste remediation. Motivated by previous work on removal of heavy metals in aqueous solution, we explored the application of germanium sulfide chalcogels as a sorbent for gas-phase I2 based on Pearson's Hard/Soft Acid–Base (HSAB) principle. This work was driven by a significant need for high-efficiency sorbents for 129I, a long-lived isotope evolved during irradiated UO2 nuclear fuel reprocessing. These chalcogel compositions are shown to possess an affinity for iodine gas, I2(g), at various concentrations in air. This affinity is attributed to a strong chemical attraction between the chalcogen and I2(g), according to the HSAB principle. The high sorption efficiency is facilitated by the high porosity as well as the exceptionally large surface area of the chalcogels. This paper briefly discusses the current and alternative waste forms for 129I, elaborates on preliminary work to evaluate a Pt-Ge-S chalcogel as a I2(g) sorbent, and discusses the unknown chalcogel properties related to these materials in waste form.
1 Introduction
The need for nuclear energy is growing in the United States to meet the necessary energy requirements for the future without the production of greenhouse emissions.1 A main concern behind nuclear power production is the control of waste products generated from nuclear fission of uranium fuel. The waste includes radioisotopes with long half-lives, t1/2, e.g., 129I (t1/2 = 1.6 × 107 yr) and 99Tc (t1/2 = 2.1 × 105 yr). These wastes must be immobilized in viable waste forms, such as glass, and then safely disposed of, usually in a geological repository.Fuel reprocessing options are currently being investigated under the U.S. Department of Energy (DOE) Office of Nuclear Energy Fuel Cycle Research and Development Program in order to recycle the reusable power-generating materials for maximum process efficiency and to potentially reduce the quantity of high-level waste, or HLW.2 One proposed reprocessing method includes a volatilization/oxidation step, commonly referred to as voloxidation,3 followed by acid dissolution and chemical separations. These techniques release volatile radionuclides within the fuel that had been generated through the nuclear fission of uranium and neutron activation of trace contaminants in the fuel and cladding, i.e., tritium (3H), carbon-14 (14C), krypton-85 (85Kr), and iodine-129 (129I).
Of these volatile radionuclides, 129I poses the greatest health risk as it has the longest t1/2 and is involved in the human metabolic process. Thus, waste containing 129I must be effectively immobilized for hundreds-of-thousands to millions of years.4 The Environmental Protection Agency (EPA) regulation 40 CFR 190 requires an 129I capture decontamination factor (DF) of 167 or capture and immobilization of >99.4 mass%.5 The decontamination factor is defined as the ratio of the original contaminant concentration to the concentration remaining following removal. To support the future expansion of nuclear energy, an effective method is required for the capture and safe storage of 129I.
Currently, the DOE plan is to use silver-exchanged mordenite, or AgZ, as the iodine sorbent for off-gas treatment at a reprocessing facility. However, the DOE is currently investigating alternative sorbents for iodine that are more efficient candidate waste forms and have higher affinity, higher waste loading capacity, and lower overall cost. Aerogels are one family of materials presently under investigation as a potential replacement for AgZ. At the Pacific Northwest National Laboratory (PNNL), we are concurrently studying two types of aerogels as iodine sorbents alternative to AgZ, (1) Ag-functionalized silica aerogels,6 and (2) unfunctionalized chalcogen-based aerogels, referred to as chalcogels.6a,6b Here, we discuss an on-going study of chalcogels as effective iodine sorbents and waste forms.
This paper discusses the following: (1) a brief summary of the current and alternative waste forms for 129I, (2) preliminary work to evaluate a Pt-Ge-S chalcogel as a I2(g) sorbent, and (3) the unknown chalcogel properties related to these materials as a potential waste form.
2 Background and principles
2.1 Iodine sorbents
Historically, various materials were investigated for the capture and immobilization of 129I. In most cases, however, the materials effective for capturing iodine gas, or I2(g), cannot subsequently be sintered and densified to create a mechanically and chemically durable waste form. [Note that 129I is used here to denote the radioactive isotope of interest and I2(g) is used to denote iodine gas, in general.] In the 1970's,7 metal-exchanged and metal-impregnated materials were evaluated as potential 129I sorbents. Among these, metal-exchanged mordenite was the most common host material investigated; mordenite is a zeolite with a nominal composition of (Ca,Na2,K2)Al2Si10O24·7(H2O), in which the alkali can be exchanged for alternative metals.In these studies,7 the alkali in the mordenite was replaced with Ag, Cd, Cu, Hg, Mn, Pb, Pd, and Tl, though only Ag proved effective at capturing I2(g). Thomas et al.7f attributed the poor I2(g) capture efficiency with metals other than silver to the fact that the metal oxides were more thermodynamically stable than the metal iodides. In the case of Ag, when comparing the Gibbs free energy of formation, ΔGf°, of Ag2Oeqn (1) and AgIeqn (2) it is evident that, thermodynamically, the formation of AgI is favored over Ag2O with a more negative ΔGf°, suggesting it is more thermodynamically preferable.
| 2Ag(c) + 0.5O2(g) → Ag2O(c) (ΔGf° = −11.28 kJ mol−1 at 298.15 K)8 | (1) |
| Ag(c) + 0.5I2(c,l,g) → AgI(c,l) (ΔGf° = −66.270 kJ mol−1 at 298.15 K)9 | (2) |
As a result of these studies, the current proposed method for removal of I2(g) from reprocessing plant process off-gases and subsequent immobilization is AgZ,4 though it has not yet been put into universal practice considering that the exact fuel reprocessing procedure is still under development. However, once online, the iodine sorbent will be packed into a sorbent bed and act as a “polishing filter” for stack gases. When AgZ is contacted with I2(g), the silver in the AgZ reacts with the I2(g) to form AgI, a very stable iodide complex. To date, AgZ has only been available as extrudates that are approximately 2 mm in diameter and 5 to 10 mm in length. This particulate form, while good for removal purposes, cannot be disposed of in a repository because of its high surface area. Thus, AgZ loaded with iodine must be further consolidated in a secondary waste form before being sent to a repository. Waste forms for encapsulating iodine-loaded AgZ include (1) a low melting glass,11 (2) cement,12 and (3) a silico-geopolymer.13 Routes for iodine removal from AgZ and post-processing include (4) apatite-like minerals, (5) low-melting glass, (6) bismuth-containing ceramics,13–14 (7) other iodide and iodate ceramics,15 and metal-organic-frameworks.16 Currently, options (1) and (2) are under development in the United States.
2.2 Silica aerogels
Functionalized silica aerogels might prove to be a suitable alternative for the AgZ primary iodine sorbent. In recent years, silica aerogels have been studied for confinement of radioactive waste.17 As with AgZ, aerogels can act as precursors to the final glass matrix in which the waste is actually immobilized.18Silica aerogels, when unmodified, generally have hydrophilic surfaces and tend to lack mechanical stability in humid environments; they become brittle when exposed to water.19 This can create difficulties when processing these materials.However, the porous network of silica aerogel has been used as a host matrix, or a sponge, for nuclear waste,19b where the silica aerogel was soaked in a solution containing actinides in nitrate salt form and, after drying and nitrate decomposition, the composite material was fully sintered, trapping the nuclear waste. Most of the problems encountered during silica aerogel studies for environmental waste remediation result from the use of aerogels in an aqueous environment, thereby exposing their inherent vulnerability to capillary forces during wetting or drying by means other than a supercritical fluid exchange.19d However, we have recently presented work demonstrating great potential for Ag-functionalized silica aerogels as potential iodine sorbents.6
2.3 Chalcogels
Chalcogels are another potential alternative to AgZ. The fundamental difference between silica aerogels and chalcogels is that the anions in chalcogels include chalcogens (Ch) S, Se, and Te but not O, whereas silica gels contain only O anions. Chalcogels can be made from an aggregation of simple binary nanocrystals (e.g., CdS, ZnS, PbS, CdSe)20 or a chemical linkage between chalcogenido clusters (e.g., Ge4S104−) and metal ions (e.g., Pt2+) that promote interlinking of the structural units.20aThe wide range of possible chalcogel compositions allows for materials engineering for a wide range of physical, chemical, and mechanical properties. Bag et al.19e and Kanatzidis and Bag21 demonstrated the compositional flexibility of (Ge,Sn)xChy chalcogels, in which properties such as specific surface area (108–323 m2 g−1) and chemical affinity could be “tuned” by changing the precursors. Bag et al. also demonstrated chalcogels as selective sorbents or catalysts for gas separation such as hydrodesulfurization.22 In addition, chalcogels can efficiently absorb organic hydrophobic aromatic molecules from solution because their hydrophobic surfaces are lined with chalcogen atoms and are physically stable at high humidity.19e Chalcogels have also been demonstrated to have a selective affinity for different ionic species in an aqueous solution; for example, Pt2Ge4S10 chalcogels have selective affinity for Hg2+ over Zn2+.19e This strongly suggests that these materials show potential as a novel platform in a wide range of environmental remediation applications.
2.4 Pearson's Hard/Soft Acid–Base principle
The selective affinity observed with the chalcogels can be explained by Pearson's Hard/Soft Acid-Base (HSAB) principle developed by Pearson and Parr.23 The HSAB principle considers the differential complexation behavior of cations and ligands in terms of electron pair-donating Lewis bases and electron pair-accepting Lewis acids.23a Pearson classified Lewis acids and Lewis bases as hard, borderline, or soft. According to Pearson's HSAB principle, hard Lewis acids prefer to bind to hard Lewis bases and weak Lewis acids prefer to bind to weak Lewis bases. Pearson classified a very wide range of atoms, ions, molecules, and molecular ions as hard, borderline or soft Lewis acids or Lewis bases.In order to explain Pearson's HSAB principle more quantitatively, one can use a parameter called chemical hardness, or η.23b Chemical hardness is proportional to the second derivative of the total energy of a chemical system (or the first derivative of the chemical potential) with respect to changes in the number of electrons in a fixed nuclear environment. The chemical hardness of atoms, molecules, or ions termed species and denoted by the subscript, s, can be calculated from the expression:
 | (3) |
| Acid/Base | Species | I s | A s | χ s | η s | Reference(s) |
|---|---|---|---|---|---|---|
| Acid | K+ | 31.63 | 4.34 | 17.99 | 13.65 | 23b,24 |
| Zn2+ | 39.7 | 17.96 | 28.8 | 10.8 | 23b | |
| HCl | 12.7 | −3.3 | 4.7 | 8.0 | 24a | |
| Hg2+ | 34.2 | 18.75 | 26.5 | 7.7 | 23b | |
| U4+ | 45.77 | 31.06 | 38.415 | 7.4 | 23b | |
| CO2(g) | 13.8 | 0 | 6.9 | 6.9 | 23b | |
| Ag+ | 21.5 | 7.57 | 14.6 | 6.9 | 23b | |
| Pu3+ | 34.6 | 21.6 | 28.1 | 6.5 | 24c | |
| Cu+ | 20.3 | 7.72 | 14.0 | 6.3 | 23b | |
| HI(g) | 10.5 | 0.0 | 5.3 | 5.3 | 24a | |
| HNO3 | 11.03 | 0.57 | 5.8 | 5.2 | 24a | |
| UO2+ | 14.6 | 6.13 | 10.365 | 4.2 | 24d | |
| I2(g) | 9.3 | 2.6 | 6.0 | 3.4 | 23b | |
| Neutral | CH3I | 9.5 | 0.2 | 4.9 | 4.7 | 24a |
| Cl2(g) | 11.6 | 2.4 | 7.0 | 4.6 | 24a | |
| Base | H2O | 12.6 | −6.4 | 3.1 | 9.5 | 24a |
| S | 10.36 | 2.08 | 6.22 | 4.12 | 23b | |
| Se | 9.75 | 2.02 | 5.89 | 3.86 | 23b | |
| Te | 9.01 | 1.97 | 5.49 | 3.52 | 23b |
The HSAB principle can be directly applied to chalcogels to obtain a qualitative understanding of the selective affinity results presented by Bag et al.19e One can postulate that the strong affinity of sulfur in a Pt2Ge4S10 chalcogel for Hg2+ over Zn2+ was a major driving factor to effectively remove Hg2+ ions from an aqueous solution. Sulfur has a chemical hardness, ηS, of 4.12, so it is considered soft and is categorized as a Lewis base. In addition, sulfur can also be classified as a soft Lewis base from its electronegativity value of 2.58; elements with an electronegativity of 2.5–3.0 are considered soft Lewis bases. The selective adsorption of Hg2+ over Zn2+ in the Pt-Ge-S chalcogel can be attributed to the fact that Hg2+ (ηHg2+ = 7.7) is a softer Lewis acid than Zn2+ (ηZn2+ = 10.8).
Manos and Kanatzidis25 reported heavy metal (i.e., Hg2+, Pb2+, and Cd2+) remediation properties of the layered sulfide material K2xMnxSn3−xS6 (x = 0.5–0.95), previously demonstrated as an excellent sorbent for strontium ions.26 Compared to other sorbents such as functionalized clays, resins, organoceramics, and mesoporous silicates with a thiol group, the layered sulfide material can be used as a sorbent without modification for various heavy metal ions. It exchanges existing intercalated K+ ions to various heavy metal ions strongly and rapidly; K+ is a well-known hard Lewis acid (ηK+ = 13.65) and various heavy metal ions are typically soft Lewis acids. This example supports the strong affinity of the sulfur atom (soft Lewis base) for soft Lewis acids.
2.5 Iodine sorption in chalcogels
Considering the chemical hardness of sulfur (ηS = 4.12, Table 1), the sulfur backbone of the Pt-Ge-S chalcogel should have a high affinity for I2(g) [ηI2(g) = 3.4]. The affinity of sulfur for I2(g) in the Pt-Ge-S chalcogel is expected to be higher than for Hg2+ as demonstrated by Bag et al.19e considering that I2(g) is an even softer Lewis acid than Hg2+ (ηHg2+ = 7.7).Other species that are present in the off-gas along with I2(g) as trace contaminants are HCl(g), HI(g), HNO3, CH3I, and Cl2(g). Of these species, two can be classified as soft Lewis acids with a chemical hardness comparable to I2(g) and those are HI(g) and HNO3 (Table 1).24aHCl is a harder Lewis acid than HI(g) [ηHCl(g) = 8.0]; ηHNO3 = 4.7 and ηCl2(g) = 4.6, although these species are considered neutral according to the HSAB principle.24a Thus, of these other species present in the off-gas stream, HI(g) and HNO3 could, potentially, compete with I2(g) for chalcogen binding sites, though the other species will, most likely, not compete according to the HSAB principle. However, since I2(g) is a weaker Lewis acid than HI(g) and HNO3, the chalcogen binding affinity for these species is predicted to be I2(g) > HNO3 > HI(g) (see Table 1).
Bearing in mind that the chemical hardness values of Se (ηSe = 3.86) and Te (ηTe = 3.52) are closer to that of I2(g), chalcogels containing these elements are expected to show an even higher affinity for iodine than compounds containing sulfur where the chalcogen affinities are predicted to be Te > Se > S. However, substituting Se and Te for S does pose drawbacks. Selenium is a toxic element controlled by the EPA under the Resource Conservation and Recovery Act27 and both selenium and tellurium-based chalcogels are often air-sensitive, making them less likely candidates for use in air. Although selenium is an essential micronutrient for animals, it is toxic and possibly cancerous at elevated levels (>400 μg day−1).28 Also it is worth noting that GeTe-based chalcogels tend to have lower specific surface areas (157–162 m2 g−1) than corresponding Ge-S (276–323 m2 g−1) and Ge-Se (282–327 m2 g−1) chalcogels.21 Thus, sulfur-based chalcogels have the preferred chemistry from a waste form standpoint because they are not as chemically toxic and/or air-sensitive as the Se and Te equivalent compounds.
3 Materials and methods
3.1 Chalcogel precursor synthesis
Many of the building blocks required for (Ge,Sn)-Ch chalcogels are unavailable commercially and were prepared hydrothermally with high-purity reactants. In order to produce R4Ge4Ch10 [R = (CH3)4N] precursors, the following chemicals were used according to methods presented in the literature:29Ge (−100 mesh, 99.9999% purity, Alfa Aesar, Ward Hill, MA), S (99.9999% purity, ASARCO, Tucson, AZ) or Se (99.9999% purity, Alfa Aesar) powders (−100 mesh), tetramethyl ammonium hydroxide pentahydrate [ROH·5H2O, Sigma-Aldrich, St. Louis, MO], and water in the molar ratios of 4, 10, 5, and 75–100, respectively, or 4 Ge![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 S:5 ROH:75 H2O. Once weighed, the chemicals were loaded into a Teflon® crucible in a Parr® acid digestion bomb (model 4748; Parr Instrument Company, Moline, IL). The reactants were stirred on a stir-plate with a Teflon-coated magnetic stir-bar while the entire vessel was heated at ∼140 °C for ≥16 h.
:10 S:5 ROH:75 H2O. Once weighed, the chemicals were loaded into a Teflon® crucible in a Parr® acid digestion bomb (model 4748; Parr Instrument Company, Moline, IL). The reactants were stirred on a stir-plate with a Teflon-coated magnetic stir-bar while the entire vessel was heated at ∼140 °C for ≥16 h.
Following the digestion, the solution was vacuum-filtered with a Büchner funnel and the filtrate collected. Ethanol was added to the filtrate to initiate precipitation of R4Ge4Ch10. In both cases, the precipitate was lightly colored, had a very small particle size, and remained suspended in the filtrate solution; the precipitate was white for Ch = S and light brown for Ch = Se. Once precipitation was visually complete, acetone was added to the solution until precipitation ceased again. In each case, the precipitate was vacuum-filtered, washed with ethanol, and then washed with acetone. The resulting precipitates were dried under vacuum and stored in an M-Braun nitrogen glovebox (M-Braun, Stratham, NH) with <0.1 ppm O2 and H2O levels to prevent oxidation or degradation. Only S-based chalcogels are discussed here.
3.2 Chalcogel synthesis
The Pt-Ge-S chalcogels were synthesized using methods outlined by Bag et al.19e,21 according to eqn (4) with the R4Ge4S10 precursors outlined above and potassium tetrachloroplatinate II (K2PtCl4, Alfa Aesar). These gels were made by combining two separate aqueous solutions of the individual precursors. For the first solution, 0.1 mmol of R4Ge4S10 (0.0908 g) was added to a 20 mL glass vial along with 3 mL of deionized water and the solution was stirred for ≥2 h at 20–25 °C. For the second solution, 0.2 mmol of K2PtCl4 (0.0830 g) was added to a separate 20 mL glass vial along with 2 mL of water and stirred for ≥2 h at 20 ± 5 °C with Teflon-coated stir-bars on a stir-plate. Being that these are non-oxide semiconducting materials, these chalcogels were assumed to be light- and/or oxygen-sensitive. Thus, both vials were wrapped in aluminium foil to prevent light exposure. After stirring the individual solutions, they were combined, stirred for 0.5–1 h, and then poured into a container where gelation [eqn (4)]occurred over a time interval that varied by experiment (Table 2).| R4Ge4S10 + 2K2PtCl4 → Pt2Ge4S10 + 4R+ + 4K+ + 8Cl− where R = (CH3)4N | (4) |
| Sample Id | Gelation time/d | Aging time/d | Degas T/°C | BET SA/m2 g−1 | BET SAeq/m2 g−1 | Pore volume/10−6 m3 g−1 | Pore size/nm |
|---|---|---|---|---|---|---|---|
| a “Gelation” and “aging” times are presented in days, “degas T” is the T/°C at which the specimen was degassed prior to making BET measurements, “BET SA” is the surface area measured in m2 g−1 (BET method), and “BET SAeq” is the SiO2 equivalent surface area calculated with eqn (5). | |||||||
| Cg-5C | 6 | 2 | 25 | 360 | 1200 | 2.3 | 20–150 |
| 60 | 342 | 1140 | 2.15 | 20–150 | |||
| 100 | 387 | 1290 | 2.38 | 20–150 | |||
| Cg-5P | 6 | 2 | 25 | 287 | 957 | 1.62 | 20–150 |
| 60 | 271 | 903 | 1.34 | 20–150 | |||
| 100 | 252 | 840 | 1.44 | 20–150 | |||
| Cg-5P+I | 6 | 2 | 25 | 48.6 | 162 | 0.32 | 20–150 |
| 60 | 83.2 | 277 | — | — | |||
| 100 | 77.6 | 259 | 0.49 | 20–150 | |||
| 125 | 66.3 | 221 | 0.43 | 20–150 | |||
| Cg-6C | 17 | 8 | 25 | 416.5 | 1388 | 2.03 | 20–150 |
| 60 | 399.0 | 1330 | — | — | |||
| 100 | 383.4 | 1278 | 2.35 | 20–150 | |||
| 125 | 355.1 | 1184 | 2.08 | 20–150 | |||
| Cg-7C | 18 | 8 | 25 | 423 | 1410 | 3.1 | 20–150 |
| 60 | 491 | 1636 | 3.4 | 20–150 | |||
| 100 | 418 | 1393 | 2.9 | 20–150 | |||
| 125 | 428 | 1426 | 3.0 | 20–150 |
We used two gel-casting techniques. The mixed solution was either poured onto a Petri dish in a fume hood19e,21—a method termed the “Plate” or “P-casting” method—or the solution was cast in a polypropylene container—a method termed the “Cylindrical” or “C-casting” method. The C-casting method was employed to improve the gelation process by eliminating the headspace above the gel. This minimized the amount of O2 available for interaction with the gel and eliminated water evaporation.
Following gelation, the chalcogels were aged, a process to increase the mechanical stability, by submersion in ethanol for 2–8 days (Table 2) at room temperature. For P-cast gels, the ethanol was simply added to the Petri dish until the gels were submersed. The C-cast gels were removed from the polypropylene containers, placed in a glass evaporation dish, and then submerged in ethanol. Following aging, gels were rinsed with water over the course of 1–2 days to remove the water-soluble products, R+, K+, and Cl− (see eqn (4)) that were not incorporated into the gel structure.
The water in the chalcogel network was then replaced with ethanol through an iterative solvent exchange process where the gels were rinsed in replenished ethanol several times over the course of 1–2 days. The ethanol was then exchanged with liquid CO2 over 8–10 rinses in a Parr bomb (model 4772(Q)). The temperature and pressure of the CO2 was increased until the critical pressure was exceeded, at which point the CO2 was released as a gas, leaving a porous and dry gel structure. Once dry, portions of each chalcogel were used for the experiments described here.
Table 2 summarizes the methods and properties of some of the chalcogels made and tested in this study. P-cast gels turned very dark in appearance after ∼24–48 h of gelation (Fig. 1) and a color gradient was observed between the top surface of the gel that was exposed to air (darker) and the bottom region in contact with the plate (lighter). The C-cast gels remained a light shade of brown even after 18 days of gelation (Fig. 2), approximately the same color as the P-cast gel between 9 and 24 h of gelation.
 | ||
| Fig. 1 Change in the appearance of P-cast Pt-Ge-S gel (Cg-3P) during the gelation process over the course of 144 h (in the hydrogel state). | ||
 | ||
| Fig. 2 Picture of Cg-7C chalcogels in a glass vial. This is the typical appearance of C-cast gels following supercritical drying. | ||
3.3 Surface area measurements
Specific surface area, porosity, and pore size were measured for most of the chalcogels with nitrogen [N2(g)] adsorption and desorption isotherms collected with a Quantachrome Autosorb-6B (Quantachrome Instruments, Boynton Beach, FL) gas sorption system on degassed samples. Samples were loaded in a glass cell and degassed at 25 °C for 16 h or at 60 °C, 100 °C, and 125 °C for 8 h while under vacuum. The degassed samples were analyzed with nitrogen adsorption and desorption at a constant temperature of 77.4 K (−195.75 °C), the temperature of liquid nitrogen. The surface area was determined from the isotherm with the Brunauer–Emmett–Teller (BET) method. The Barrett–Joyner–Halenda (BJH) method was used for the porosity and pore size analyses.Since the elements used to prepare chalcogels are much heavier than those for silica aerogels, a direct comparison of specific surface areas does not adequately portray the available surface area. To compare the surface area of the chalcogels to silica aerogels, the most common aerogel compound, Bag et al.19e,21 developed a SiO2 surface area equivalent, or SAeq. This SAeq value is simply a way of normalizing the chalcogel composition to two anions, which in this case, are chalcogen atoms, i.e., Pt0.4Ge0.8S2 for the Pt2Ge4S10 chalcogel. The value of SAeq can be calculated from the following expression:
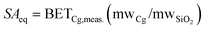 | (5) |
3.4 X-Ray diffraction
Portions of each precipitate were ground to powder in an agate mortar and pestle and analyzed with X-ray diffraction (XRD) to verify the phases present. The analysis was performed with a Bruker® D8 Advance (Bruker AXS Inc., Madison, WI) equipped with a Cu-Kα target at 40 kV and 40 mA. The instrument had a LynxEyeTM position-sensitive detector with an angular range of 3° 2θ. Scan parameters used for sample analysis were 5–110° 2θ with a step of 0.015° 2θ and a 0.3 s dwell at each step. JADE 6© (Materials Data, Inc., Livermore, CA), Bruker AXS DIFFRACplusEVA, and Bruker AXS Topas 4.2 software were used to identify and quantify phase assemblages. Both S- and Se-based precipitates matched the diffraction pattern for R4Ge4S10 presented by Pivanet al.,30 with a powder diffraction file (PDF) number 52-7775, after small sample displacement adjustments.The samples Cg-5P and Cg-5P+I (see Table 2) were prepared for XRD analysis after being in a vacuum desiccator for several months following the completion of gelation and iodine sorption experiments. The gels were suspended in ethanol within small glass vials and were crushed to a fine powder. Each suspension was added drop-wise onto a separate zero-background silicon wafer XRD sample holder, the ethanol was allowed to evaporate, and then the samples were analyzed using the same parameters as mentioned above.
3.5 Iodine uptake experiments
The effect of P- and C-casting methods on maximum iodine sorption of chalcogels was studied with chalcogels Cg-5P and Cg-5C, respectively. Each specimen was loaded into a separate pre-weighed glass vial and loaded into a vacuum desiccator with 99.9999% pure, nonradioactive iodine (Alfa Aesar, Ward Hill, MA) at a pressure of 1.3–6.7 Pa (10–50 mTorr) and removed periodically for mass measurements, which were recorded to the nearest 0.1 mg on a Mettler AE200 analytical balance (Mettler-Toledo, Inc., Columbus, OH). Initial masses of the Cg-5C and Cg-5P were 0.0465 g and 0.1032 g, respectively. Following the maximum sorption experiment, the iodine was removed from the desiccator and both chalcogels were left in the desiccator under vacuum to facilitate desorption of physisorbed I2(g). Sorption values were then normalized to the iodine uptake of an empty glass vial, identical to the vials used to contain the samples.A separate I2(g) sorption test was performed in low iodine concentrations and elevated temperatures (∼140 °C), closer aligned to a simulated waste stream environment than an idealized, saturated iodine atmosphere. For this experiment, 0.0666 g of Cg-7C was packed lightly into a 10 mL glass pipette (occupied ∼1.3 mL with ρbulk = 51.2 kg m−3) and held vertical with a porous silica disc (Fig. 4-A) while a dilute stream of iodine was allowed to flow through the sorbent column.
In order to achieve a steady flow of iodine for this experiment, we passed a stream of dry air through a DYNACAL®iodine permeation tube with a permeation rate of 22.8597 ng s−1, resulting in a concentration of 4.2 ppm I2(g) by volume. The gas passed through the pipette to a bubbler where it was scrubbed in 0.1 M NaOH solution that was changed every 30 min. A schematic of the experimental setup is presented elsewhere.6b
In order to calibrate the flow of iodine through the system, a blank run was made with no specimen in the pipette. After 30 and 60 min, the scrubber solutions were changed and the dissolved iodine concentration was measured. The concentrations were 1.234 and 2.495 g m−3, respectively, or an I2(g) flow rate of 691.5 μg m−3s−1. During the experiment with the chalcogel sorbent, the pipette with Cg-7C was inserted into the air/I2(g) stream (Fig. 4-A). The 0.1 M NaOH scrubber solutions were removed at 1.28 × 103, 1.10 × 104, and 1.26 × 104 s during the sorption process and were analyzed by inductively coupled plasma mass spectrometry (ICP-MS).
3.6 Thermal analysis
Chalcogels with (Cg-5C+I) and without (Cg-7C) sorbed iodine were analyzed with a Seiko thermogravimetric differential thermal analyzer (TGA-DTA) in platinum pans without lids. Prior to the analysis, the iodine content of Cg-5C+I was verified by scanning electron microscopy and energy dispersive X-ray spectroscopy (SEM-EDS) to be 39 ± 2 mass%, with the variation depending on the analysis region. The specimens were heated from room temperature to 800 °C at 0.0833 °C s−1 and the cool-down curves were disregarded.3.7 Scanning electron microscopy and energy dispersive spectroscopy
Chalcogel microstructures and compositions were analyzed on select samples with two instruments. A JEOL JSM-5900 (JEOL Ltd., Tokyo, Japan) SEM with a tungsten filament was used with an EDAX Si-drifted EDS detector (AMETEK, Berwyn, PA). Additionally, sorbed iodine in select gels and the Pt:Ge:S ratios in the as-dried gels were analyzed with this EDS detector.
A second JEOL SEM, a JSM-7001FTTLS model with a field emission gun, was used to collect higher resolution micrographs of select chalcogels. This instrument was operated in high vacuum mode on an uncoated specimen with a 3 kV acceleration voltage, a 500 V stage bias, and a 4 mm working distance. Micrographs were captured with an in-lens upper electron detector (UED) operated in backscattered mode.
4 Results and discussion
4.1 Iodine sorption experiments
The primary goal of this work was to demonstrate the effectiveness of Pt-Ge-S chalcogels as a sorbent for I2(g) in dilute streams. Here, we have shown that Pt-Ge-S chalcogels have a high affinity and capacity for iodine in both saturated (Fig. 3) and low (Fig. 4) iodine concentration environments. The mass of iodine sorbed on the empty vial (blank) used for the maximum iodine sorption experiments was very low (<0.002 g); however, this value was used to renormalize values of the iodine sorbed on the chalcogels. A very high I2(g) sorption (196%) was demonstrated in Cg-5C after ∼5 days in the vacuum desiccator and 239% by mass after 20.0 days (Fig. 3). The desorption of I2(g) was found to be very low compared to the rate of adsorption and appeared to be linear over the course of ∼25 days (Fig. 3). These experiments were done for demonstration purposes only as a comparison between the gels made with different casting techniques. Efforts to reproduce these results were not deemed necessary considering that the actual sorption environment will be far from a saturated iodine concentration. | ||
| Fig. 3 Comparison plot of iodine sorption (mass%) between P-cast and C-cast gels over time in a vacuum desiccator. “Adsorption” was terminated after 20 days when the iodine source was removed from the vacuum desiccator at which point “desorption” began as the gels remained under vacuum. | ||
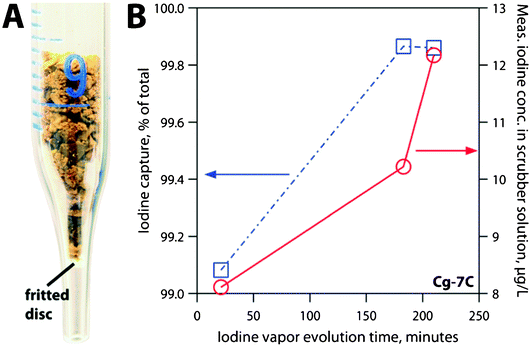 | ||
| Fig. 4 (A) Iodine sorption experiment (showing low packing density) and (B) results of iodine capture in terms of % of total iodine (from calibration run) and measured iodine concentration in the scrubber solution (μg L−1) on specimen Cg-7C. | ||
The amount of sorbed I2(g) in Cg-5C (239 mass%) was higher than that of Cg-5P (87.1 mass%). The difference can most likely be attributed to the higher specific surface area measured for Cg-5C (360 m2 g−1) as compared to Cg-5P (287 m2 g−1) (Table 2). Following the I2(g) saturation experiment, the surface area for Cg-5P+I degassed at 25 °C was found to be 48.6 m2 g−1, compared to 287 m2 g−1 for Cg-5P, and the pore volume to be 3.2 × 10−7 m3 g−1, compared to 1.6 × 10−6 m3 g−1 in Cg-5P. Both values represent a large decrease from the values for Cg-5P, which was expected due to the amount of iodine present.
A color gradient was observed across chalcogels during early iodine sorption scoping studies in the vacuum desiccator, where the outside of the chalcogel granules appeared darker than the interior regions. This suggests that the iodine was preferentially adsorbing to the outer, more exposed surfaces of the chalcogel granules. No compositional analysis was performed on these gels to determine the iodine concentration gradient.
The chalcogel also performed very well in the low concentration iodine sorption experiment. The pipette containing the iodine-sorbed gel was weighed after the experiment and the final mass increase was determined to be 0.0012 g (1.80% increase for the chalcogel, by mass). Even with the relatively large granules and low packing density (Fig. 4-A), >99% I2(g) sorption (Fig. 4-B) was found for the P-cast chalcogels over the entire duration of the experiment. The NaOH scrubber solutions collected at 1.28 × 103, 1.10 × 104, and 1.26 × 104 s showed iodine concentrations of 8.11 × 103, 10.2 × 103, and 12.2 × 103 μg m−3, which equate to DFs of 109, 743, and 716, respectively. The first collection point at 1.28 × 103 s showed a much higher dissolved concentration of iodine than expected and was inconsistent with the other values. However, the latter two data points clearly demonstrate the potential of these materials to remove even dilute iodine from an air stream, with very good DFs. Future experiments will be performed with either powdered chalcogels or smaller granules to allow greater interaction between the gas stream and the interior chalcogel surfaces.
4.2 Gelation method comparisons
The increase in specific surface area and iodine capacity in the C-cast gels compared to the P-cast gels (Table 2) is attributed to the reduction in oxygen exposure as well as to the elimination of light exposure and liquid evaporation potential with the C-casting method. The P-cast gels seen in Fig. 1 decreased in height during gelation, although the contact area on the plate remained constant, resulting in an overall volumetric decrease. The C-cast gels (Fig. 2), even after several weeks inside the polypropylene vial did not shrink at all and were actually difficult to remove from the vial. This suggests that the C-casting method is more effective at preserving the pore structure of the gel during the gelation process, resulting in noticeably higher overall specific surface areas for C-cast gels.The P-cast gels had surface areas consistent with, but slightly lower (∼10%) than, those reported in the literature, i.e., 252–287 m2 g−1versus 276–323 m2 g−1 at 25 °C (Table 2).19e,21Cg-7C, a C-cast gel, had a much higher specific surface area than the P-cast gels, with a maximum value after degassing at 60 °C of 491 m2 g−1. This value can be compared to literature values of ∼300 m2 g−1 after degassing at 75 °C.19e,21 The corresponding SAeq for Cg-7C at this maximum value is 1636 m2 g−1 (Table 2).
The N2(g) adsorption/desorption isotherms appear similar for C-cast, P-cast gel and P-cast gel with sorbed iodine without considering the volume scale differences (Fig. 5). The isotherms appear to be consistent with Brunauer Type II isotherms,31 representing multilayer adsorption of N2(g) during the BET measurement, and moderate hysteresis was observed. Note that the isotherm shown in Fig. 5-C demonstrates a dramatic difference in the N2(g) adsorption capacity, compared to the other two gels (Fig. 5-A and Fig. 5-B), because of the adsorbed iodine.
 | ||
| Fig. 5 BET isotherms for (A) Cg-5C, (B) Cg-5P, and (C) Cg-5P+I collected after degassing at 25 °C for 16 h. Adsorption (○) and desorption (□) are presented as separate datasets. | ||
During iodine sorption experiments, the chalcogels were not powdered, thus reducing the packing density in the test vial. A color gradient was observed on a cross-section of the C-cast gels following the iodine sorption experiments where the outer rim appeared dark blue while the interior remained light brown in color. This observation suggested that the iodine did not penetrate into the gel interior during these uptake experiments as effectively as if the gel had been in powder form, fully exposing the inner porosity of the chalcogels. This implies that the sorption efficiency would potentially increase with a higher chalcogel packing density.
4.3 X-Ray diffraction
The XRD patterns of Cg-2P and Cg-3P were amorphous with minor diffraction peaks partially matching Ge (PDF#72-1089)32 and GeO2 (PDF#34-1089)33 (Fig. 6-A). The diffraction peaks observed with XRD for Cg-5P and Cg-5P+I match those of GeO2 (PDF# 85-0473),34 suggesting that the darker color of aged Pt-Ge-S chalcogels can possibly be attributed to the oxidation of free germanium in the chalcogel (Fig. 6-B), according to the reaction (see Fig. 6-C):| Ge(c,l) + O2(g) → GeO2(c) (ΔGf°= −521.339 kJ mol−1 at 298.15 K)8 | (6) |
 | ||
| Fig. 6 XRD patterns of (A) Cg-2P and Cg-3P showing amorphous structure with minor diffraction peaks partially matching Ge (PDF#72-1089,32 □) and GeO2 (PDF#34-1089,33 ○). (B) XRD patterns of Cg-5P, Cg-5P + iodine aged in air matching an XRD pattern for GeO2 (PDF# 85-0473,34 •). (C) Backscattered electron (SEM) micrograph of Cg-5P aged in air showing the darker, faceted, oxidized/crystallized regions. | ||
In Fig. 6-B, the color gradient from a bright orange to a dark brown/black in Cg-5P can be clearly seen. This gradient is caused by air exposure of part of the gel during the aging and solvent exchanges, a phenomenon which could probably be eliminated with the C-casting process or by P-casting in an inert atmosphere. The link of the dark color to air exposure is supported by the observation of a lighter brown color of the region of gel against the Petri dish and less exposed to air or to which air had not diffused sufficiently.
4.4 Thermal analysis
Results from the TGA for the chalcogel without iodine, Cg-7C, are presented in Fig. 7-A. They show a mass loss of 55–60 mass% after heating to 800 °C. Results from the TGA for the iodine-sorbed chalcogel, Cg-5C+I, are shown in Fig. 7-B. They shows a high rate of mass loss at T ≥ 100 °C (∼60% loss by mass at 200 °C), which is attributed to iodine desorption and decomposition of the chalcogel. It is worth noting that these chalcogels did not appear to melt at these temperatures. | ||
| Fig. 7 Thermogravimetric analysis (TGA) and differential thermal analysis (DTA) on (A) Cg-7C and (B) Cg-5C+I (with 38–41 mass% iodine). | ||
The Pt-Ge-S chalcogels do not have high thermal stabilities, especially at temperatures in excess of 300–400 °C, which will be required for consolidation following iodine sorption. In the TGA experiments, the mass loss in the gel without iodine, Cg-7C, was very comparable to data presented by Bag et al.19e for Pt2Ge4S9.6. Bag et al. attribute the mass loss at T = 25–180 °C to physisorbed or chemisorbed water or ethanol, residual from the solvent exchange and supercritical drying process. The mass loss from 180–440 °C was attributed to the loss of four sulfur atoms (thermal decomposition of the gel) and the mass loss at T > 550 °C is attributed to further thermal decomposition of the gel. The thermal stability of the iodine-sorbed chalcogel, Cg-5C+I, at T ≳ 150 °C (Fig. 7-B) was attributed to significant thermal decomposition of the gel. These decomposition products will likely be verified in future experiments with a mass spectrometer coupled to a simultaneous thermal analyzer.
Achieving thermal stability in a waste form for iodine is essential to reducing the likelihood of phase changes or iodine desorption during consolidation and immobilization. Achieving high waste-loading in a Pt-Ge-S chalcogel while maintaining temperature stability might prove to be problematic but this can likely be achieved with changes in the chalcogel chemistry. For example Bag et al.19e demonstrated a lower mass loss with Pt-Ge-Se gels over Pt-Ge-S chalcogels (loss of 30% of initial mass compared to ∼38%, respectively, after heating to 800 °C).
Recent work by Oh et al.35 with Zn-Sn-S and by Bag and Kanatzidis36 with Sn-S chalcogels showed better thermal stability with >88% and >92% mass retention after heating to 600 °C, respectively. These alternative gel chemistries seem like they could hold more promise than the Ge-S gels from a thermal stability standpoint.
5 Application of chalcogels as a waste form
The general approach towards implementing chalcogels as a waste form will require a formulation that meets the requirements, e.g., high iodine affinity, chemical durability, and thermal stability. Glasses and glass ceramics are common waste form materials because they provide the advantage of compositional flexibility. The compositions of these materials can be tailored to produce properties suitable for various types of wastes, such as viscosity, melting temperature, thermal stability, and chemical durability; however, often improving one of these properties is detrimental for another.Conveniently, chalcogels are composed of the constituents required to make a unique type of glass called a chalcogenide glass. These glasses can be made by combining at least one chalcogen ion, excluding O and Po, and at least one more electropositive element, e.g., As, Sb, Ge. A wide range of chalcogenides can be made into glass and it is possible to incorporate large quantities of iodine into the structure of these chalcogenide glasses.37 For example, the iodine solubility limit in germanium sulfide chalcogenide glass was demonstrated at >70 mass% (23.6, 2.1, and 74.31 mass% of Ge, S, and I, respectively).37i This indicates promise for melt-processed chalcogenide glass waste forms with high iodine-loading. This provides an opportunity to immobilize iodine-sorbed chalcogels by consolidating them into a chalcogenide glass at low-to-moderate temperatures (≤800 °C).
Chalcogenide glasses are typically made by sealing the elemental components in an evacuated fused quartz ampoule to isolate them from oxygen during heating. Heating material in a sealed quartz container does pose safety concerns, especially if the materials contain radioactive isotopes, however alternate methods would be implemented for processing of actual waste. For example, halide-containing chalcogenide glasses can be processed outside of this regime in covered crucibles.37h One drawback to chalcogenide glasses is that there are few published composition–property relationships and models for these materials, especially those pertaining to the waste form properties of these glasses.
5.1 Chemical durability
From a waste form standpoint, the targeted chalcogel and chalcogenide glass compositions should be chemically durable. However, not much is known about the chemical durability of chalcogenide glasses under standard conditions, e.g., with an approved standard glass leaching procedure like the Product Consistency Test38 that is often used to evaluate borosilicate waste glasses for radioactive waste remediation. However, chalcogenide glasses are compositionally flexible and thus can likely be engineered with desirable chemical durability. According to Popescu,39 “The amorphous chalcogenides are chemically stable materials in normal conditions...they are not greatly affected by the aggressive media, are not soluble in water, and do not dissolve in organic solvents.” It is well-known that amorphous chalcogenide glasses are not chemically stable in alkaline solutions.40It is important to consider the chemical durability of chalcogels when considering the harsh environment that sorbents will be subjected to in the off-gas stream. As already discussed, the off-gas stream contains H2O and HNO3; most likely these will both be in the vapor phase. Conveniently, most chalcogenides are acid-resistant so this might prove to be a benefit. Also, the chalcogels appear to be very stable in aqueous solutions35 so that is a benefit. However, the possibility exists that these off-gas components could oxidize the chalcogel and that will need to be investigated in the future.
The chemical stability of the chalcogenide glasses increases with the increase of the degree of metallization of the bonds in the series S → Se → Te.41 This is supported by the increase in the activation energy of dissolution and of the alkali concentration wherein the glass dissolution takes place. The elements of Group IV-A of the periodic table of the elements (i.e., Si, Ge) increase the stability of the glass by the formation of tetrahedral units (e.g., GeCh4/2, GeAsCh4/2).42 This information related to chalcogenide glasses can be extrapolated to chalcogels in order to engineer a waste form with high chemical durability. However, in order to improve the chemical durability of the chalcogenide glasses, the dissolution mechanism will need to be investigated further.
5.2 Thermal stability
The targeted chalcogenide glass would have to be thermally stable. Glass undergoes a key structural phase change at the glass transition temperature, Tg. If a glass is heated above Tg, it can potentially undergo crystallization at the crystallization temperature, Tc, resulting in additional mechanical stresses in the glass network from the thermal expansion mismatch between the amorphous and crystalline phases. The Tg and Tc can be measured with DTA and the ΔT, Tc − Tg, can be determined in order to help evaluate the tendency of a particular glass to crystallize. This evaluation is often referred to as the Hruby criterion43 and Tc refers to the peak temperature of crystallization. A variation of the Hruby is preferred where the temperature of the onset of Tc, or Tx, is used instead (i.e., Tx − Tg) to evaluate resistance to devitrification.44With that in mind, iodine addition to a Ge-S chalcogenide glass has been demonstrated to lower Tg significantly.37a–37g This is both a benefit and a disadvantage. It is a benefit because a lower Tg leads to a lower melting point, that allows for lower temperature consolidation. It could be a disadvantage because it will decrease the thermal stability. One concern with radioactive materials as the radionuclide concentration increases is the temperature that the waste form achieves because of the radiodecay heat. In the case of 129I, the heat generation is quite low, 1.288 × 10−8 W kg−1. Thus, even a chalcogenide glass containing the maximum I2 loading would not contain enough 129I to generate much heat. Assuming that all the iodine is 129I, a canister containing 3000 kg of glass or 1500 kg of 129I2 would generate approximately 0.02 mW of heat. Therefore, the storage temperature of a fully loaded chalcogel should be essentially ambient. This suggests that the effect on thermal stability for iodine-loaded chalcogels will likely not be a key issue of concern.
Considering the Ge-S-I ternary chalcogenide glass system, a least squares model for Tg based on literature data37a–37g reveals partial specific Tg coefficients of 524.2 °C, 173.3 °C, and 73.37 °C for Ge, S, and I, respectively, for compositions based in mass fractions; these coefficients show the effect of each component on Tg, individually. The predicted compared with the measured values for this model are presented in Fig. 8. According to this model, a Ge4S10 (Ge29S71) glass without iodine should have a Tg of 340 °C. A glass made from Ge4S10 chalcogel loaded with 30 mass% iodine would have a Tg of 260 °C. With this model, the Tg of a Ge-S chalcogenide glass can be predicted at different iodine loadings. However, an interlinking metal is required to make a Ge-S chalcogel and this additive changes the thermal properties of the consolidated waste form. The Tc of these glasses would also have to be taken into consideration to determine the glass formation ability with the Hruby criterion.43
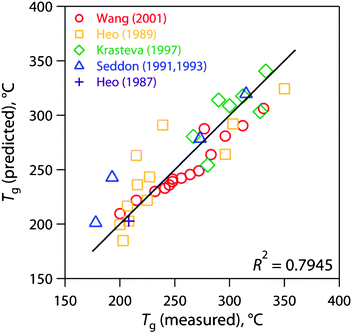 | ||
| Fig. 8 Glass transition temperature (Tg) model based on the Ge-S-I ternary system for chalcogenide glasses incorporating data from the literature (38 data points).37a–37g | ||
5.3 AgI incorporation
Silver mordenites were developed, in part, because of the strong AgI bond.7,10 Conveniently, silver iodide is very soluble in chalcogenide glasses.45 Thus, Ag could be introduced into the gel either as part of the structure or simply as nanoparticles adsorbed onto the internal pore structure of the gel. One potential method for introducing Ag into the chalcogel network is with Ag-containing precursors, e.g., R2Ag2Ge4S10 in eqn (4),29 although this has not yet been demonstrated. This material could then be used to remove I2(g) from the process off-gases.Once the AgI complex is formed in the chalcogel, it can then be immobilized as a chalcogenide glass. In particular, AgI solubility in Ge-S45c and Ge-Se45d,45echalcogenide glasses has been studied where up to 87 mass% and 62 mass% of AgI, respectively, were successfully incorporated into glasses. In one of these studies, Vassilev et al.45e demonstrated high AgI solubility (≤22 mass%) in a Zn-Ge-Se glass, the target glass system for Ge-Se-based precursors with a zinc interlinking metal. Zn-based precursors (e.g., Zn acetylacetonate35) will provide lower cost alternatives to Pt-based precursors.
6 Summary and conclusions
Chalcogels were predicted to be an effective I2(g) sorbent with Pearson's HSAB principle based on the affinity of a soft Lewis base such as a chalcogen (i.e., S, Se, and Te) for a soft Lewis acid such as I2(g). In order to test this theory, Pt-Ge-S chalcogels with high specific surface areas (up to 490 m2 g−1) were made with a technique where the gels were cast in cylindrical polyethylene containers with zero headspace (C-cast method) to reduce oxidation and solvent evaporation during gelation. The C-cast gels had up to ∼60% increase in surface area compared to similar gels presented in the literature (∼300 m2 g−1), a difference attributed to the different casting method and the longer gelation times than those presented in the literature.High I2(g) affinity was demonstrated with the Pt-Ge-S chalcogels, both under a saturated iodine environment and gas containing low I2(g) concentrations. The maximum iodine loading was about 240 mass%. More than 99% of the available I2 was removed from flowing dry air containing I2(g) at 4.2 ppm. We observed thermal decomposition when heating the iodine-sorbed chalcogels. The Pt-Ge-S chalcogels do not melt at T ≤ 800 °C. These results indicate that significant improvements to the chalcogels are needed to ensure a strong iodine chemisorption and prevent desorption during consolidation. To make chalcogels viable for use in a reprocessing plant to remove I2(g) from the process off-gases additional work is needed including (1) changing gel chemistry to achieve better thermal stability (e.g., substituting Zn for Pt, Sn for Ge, Se for S) or (2) adding strong iodine binding materials into the chalcogel network with alternate precursors or as a surface coating of nanoparticles (e.g., Ag).
More work is still needed to better understand the composition–property relationships in these materials. One of the most important composition–property relationships to further explore is chemical durability. Determining the chalcogenide glass compositions with high chemical durability will be an important step towards engineering environmentally stable waste forms out of these materials.
Acknowledgements
Pacific Northwest National Laboratory (PNNL) is operated for the U.S. Department of Energy by Battelle under Contract DE-AC05-76RL01830. This work was conducted under the U.S. Department of Energy Office of Nuclear Energy Fuel Cycle Research and Development program. The authors would like to thank Y. Su for initial help with BET measurements, A. E. Kozelisky for performing thermal analysis, J. S. McCloy for helpful comments on the manuscript, and L. Buchanan for project management support. The authors would also like to recognize R. T. Jubin for his overseeing of this project. Also, the authors extend special thanks to M. G. Kanatzidis and S. Bag at Northwestern University for helpful discussions and Naoki Kikuchi at JEOL Ltd. for providing difficult-to-obtain SEM micrographs of the uncoated chalcogels. The graphical abstract was modified from Stephanie Brock's schematic of Hg2+ capture on Pt-Sn-Se chalcogels (Science, 2007, 317, 460).References
- J. Vienna, Int. J. Appl. Glass Sci., 2010, 1, 309 Search PubMed.
- (a) J. Carter, A. Cozzi, R. Jones, G. Matthern , M. Nutt, S. Priebe and K. Sorenson, Global Nuclear Energy Partnership Integrated Waste Management Strategy, GNEP-WAST-WAST-AI-RT-2008-000214, Savannah River National Laboratory, Aiken, SC, 2008 Search PubMed; (b) Nuclear Energy Research and Development Roadmap, U.S. Department of Energy, Nuclear Energy, Washington, D.C., 2010 Search PubMed.
- R. Wigeland, T. Bjornard and B. Castle, The Concept of Goals-Driven Safeguards, INL/EXT-09-15511, Idaho National Laboratory, Idaho Falls, ID, 2009 Search PubMed.
- K. W. Chapman, P. J. Chupas and T. M. Nenoff, J. Am. Chem. Soc., 2010, 132, 8897 CrossRef CAS.
- R. T. Jubin, G. D. DelCul, B. D. Patton, R. S. Owens, D. W. Ramey and B. B. Spencer, in Proc. of the Waste Management, 2009, pp. 1–13 Search PubMed.
- (a) J. V. Ryan, E. C. Buck, J. Chun, J. V. Crum, B. J. Riley, D. M. Strachan, S. K. Sundaram, L. A. Turo and J. D. Vienna, Alternate Waste Forms: Aqueous Processing, AFCI-WAST-PMO-MI-DV-2009-000360, PNNL, Richland, WA, 2009 Search PubMed; (b) D. Strachan, J. Chun, C. H. Henager Jr, J. Matyas, B. J. Riley, J. V. Ryan and P. K. Thallapally, Summary Report for the Development of Materials for Volatile Radionuclides, PNNL-20007, Pacific Northwest National Laboratory, Richland, WA, 2010 Search PubMed; (c) J. Matyáš, G. E. Fryxell, B. J. Busche, K. Wallace and L. S. Fifield, Functionalized Silica Aerogels: Advanced Materials to Capture and Immobilize Radioactive Iodine, in Proceedings of the Ceramic Engineering and Science: Ceramic Materials for Energy Applications, ed. H. Lin, Y. Katoh, K. M. Fox, I. Belharouak, S. Widjaja and D. Singh, Volume 32, Issue 9, pp. 23–33, 2011 Search PubMed.
- (a) D. T. Pence, F. A. Duce and W. J. Maeck, in Proc. of the 11th AEC Air Cleaning Conference, CONF 700816, 2, 1970 Search PubMed; (b) W. J. Maeck and D. T. Pence, in Proc. of the 11th AEC Air Cleaning Conference, CONF-700816, 2, 1970 Search PubMed; (c) R. D. Ackley and Z. Combs, Applicability of Inorganic Sorbents for Trapping Radioiodine from LMFBR Fuel Reprocessing Off-Gas, ORNL/TM-4227, Oak Ridge National Laboratory, Oak Ridge, TN, 1973 Search PubMed; (d) I. J. Gal, A. Muk, M. Todorovic, J. Rajnvajn, D. Cvjeticanin, L. Vujisic and M. W. First, in Proc. of the 13th AEC Air Cleaning Conference, CONF 740807, 1974 Search PubMed; (e) D. T. Pence, F. A. Duce and W. J. Maeck, in Proc. of the 12th AEC Air Cleaning Conference, CONF 720823, 1972 Search PubMed; (f) T. R. Thomas, L. P. Murphy, B. A. Staples and J. T. Nichols, Airborne elemental iodine ioading capacities of metal zeolites and a method for recycling silver zeolite, ICP-1119, Idaho National Laboratory, Idaho Falls, ID, 1977 Search PubMed.
- L. B. Pankratz, Thermodynamic properties of elements and oxides, U.S. Department of the Interior, Bureau of Mines, Washington, D.C., 1982, vol. 672 Search PubMed.
- L. B. Pankratz, Thermodynamic properties of halides, U.S. Department of the Interior, Bureau of Mines, Washington, D.C., 1984, vol. 674 Search PubMed.
- D. R. Haefner and T. J. Tranter, Methods of Gas Phase Capture of Iodine from Fuel Reprocessing Off-Gas: A Literature Survey, INL/EXT-07-12299, Idaho National Laboratory, Idaho Falls, ID, 2007 Search PubMed.
- (a) T. M. Nenoff, J. L. Krumhansl, T. Garino, N. W. Ockwig and K. Holt-Larese, Waste Form Development and Testing for I2, GNEP-WAST-PMO-MI-DV-2008-000149, U.S. Department of Energy, Office of Nuclear Energy, Washington, D.C., 2008 Search PubMed; (b) T. J. Garino, T. M. Nenoff, J. L. Krumhansl and D. X. Rademacher, J. Am. Ceram. Soc., 2011, 94, 2412 CrossRef CAS.
- R. D. Scheele, C. F. Wend, W. C. Buchmiller, A. E. Kozelisky and R. L. Sell, Preliminary Evaluation of Spent Silver Mordenite Disposal Forms Resulting from Gaseous Radioiodine Control at Hanford's Waste Treatment Plant, WTP-RPT-039, Battelle - Pacific Northwest Division, Richland, WA, 2002 Search PubMed.
- J. D. Vienna, Waste Forms for an Advanced Fuel Cycle, http://www.cresp.org/NuclearChemCourse/monographs/, 2009 Search PubMed.
- J. L. Krumhansl and T. M. Nenoff, Appl. Geochem., 2011, 26, 57 CrossRef CAS.
- (a) M. Barnes, D. Roy and L. Wakeley, Leaching of composites of cement with the radioactive waste forms strontium powellite and iodine sodalite at 90 °C, in Advances in Ceramics, ed. G. Wicks and W. Ross, American Ceramic Society, Columbus, OH, 1983, vol. 8, pp. 406–412 Search PubMed; (b) D. M. Strachan and H. Babad, Am. Ceram. Soc. Bull., 1979, 58, 327; (c) E. Vance, W. B. White and R. Roy, Am. Ceram. Soc. Bull., 1980, 59, 397.
- (a) D. F. Sava, M. A. Rodriguez, K. W. Chapman, P. J. Chupas, J. A. Greathouse, P. S. Crozier and T. M. Nenoff, J. Am. Chem. Soc., 2011, 133, 12398 CrossRef CAS; (b) D. F. Sava, T. J. Garino and T. M. Nenoff, Ind. Eng. Chem. Res., 2011 DOI:10.1021/ie200248g.
- (a) S. J. Coleman, P. R. Coronado, R. S. Maxwell and J. G. Reynolds, Environ. Sci. Technol., 2003, 37, 2286 CrossRef CAS; (b) E. Sizgek, J. R. Bartlett and M. P. Brungs, J. Sol-Gel Sci. Technol., 1998, 13, 1011 CrossRef CAS; (c) M. Toki, S. Miyashita, T. Takeuchi, S. Kanbe and A. Kochi, J. Non-Cryst. Solids, 1988, 100, 479 CrossRef CAS; (d) E. R. Vance, J. Mater. Sci., 1986, 21, 1413 CrossRef CAS; (e) T. Woignier, J. Reynes, J. Phalippou, J. L. Dussossoy and N. N. Jacquet-Francillon, J. Non-Cryst. Solids, 1998, 225, 353 CrossRef CAS.
- T. Woignier, J. Reynes, J. Phalippou and J. L. Dussossoy, J. Sol-Gel Sci. Technol., 2000, 19, 833 CrossRef CAS.
- (a) M. Aparicio, M. O. Prado and A. Durán, J. Non-Cryst. Solids, 2006, 352, 3478 CrossRef CAS; (b) J. Reynes, T. Woignier and J. Phalippou, J. Non-Cryst. Solids, 2001, 285, 323 CrossRef CAS; (c) G. W. Scherer, H. Hdach and J. Phalippou, J. Non-Cryst. Solids, 1991, 130, 157 CrossRef CAS; (d) T. Woignier, J. Primera, M. Lamy, C. Fehr, E. Anglaret, R. Sempere and J. Phalippou, J. Non-Cryst. Solids, 2004, 350, 299 CrossRef CAS; (e) S. Bag, P. N. Trikalitis, P. J. Chupas, G. S. Armatas and M. G. Kanatzidis, Science, 2007, 317, 490 CrossRef CAS.
- (a) J. L. Mohanan, I. U. Arachchige and S. L. Brock, Science, 2005, 307, 397 CAS; (b) K. K. Kalebaila, D. G. Georgiev and S. L. Brock, J. Non-Cryst. Solids, 2006, 352, 232 CrossRef CAS.
- M. G. Kanatzidis and S. Bag, Semiconducting aerogels from chalcogenido clusters with broad applications, Patent application number US 2008/0241050 A1, 2008 Search PubMed.
- S. Bag, A. F. Gaudette, M. E. Bussell and M. G. Kanatzidis, Nat. Chem., 2009, 1, 217 CrossRef CAS.
- (a) R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533 CrossRef CAS; (b) R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512 CrossRef CAS.
- (a) R. G. Pearson, Chemical Hardness, Wiley-VCH Verlag GmbH, Weinheim, Germany, 1997 Search PubMed; (b) N. C. Pyper and I. P. Grant, J. Chem. Soc., Faraday Trans. 2, 1978, 74, 1885 RSC; (c) S. Cotton, Lanthanide and Actinide Chemistry, Inorganic Chemistry: A Textbook Series, John Wiley & Sons, Ltd, West Sussex, 2006 Search PubMed; (d) J. K. Gibson, R. G. Haire, J. Marçalo, M. Santos, A. Pires de Matos and J. Paulo Leal, J. Nucl. Mater., 2005, 344, 24 CrossRef CAS; (e) J. F. Liebman, J. Chem. Educ., 1973, 50, 831 CrossRef CAS.
- M. J. Manos and M. G. Kanatzidis, Chem.–Eur. J., 2009, 15, 4779 CrossRef CAS.
- M. J. Manos, N. Ding and M. G. Kanatzidis, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3696 CrossRef CAS.
- Environmental Protection Agency, Federal Regulation 40 CFR 261.
- Selenium, in Dietary Reference Intakes for Vitamin C, Vitamin E, Selenium, and Carotenoids, Food and Nutrition Board, Institute of Medicine, 2000 Search PubMed.
- C. L. Bowes, W. U. Huynh, S. J. Kirkby, A. Malek, G. A. Ozin, S. Petrov, M. Twardowski and D. Young, Chem. Mater., 1996, 8, 2147 CrossRef CAS.
- J. Y. Pivan, O. Achak, M. Louër and D. Louër, Chem. Mater., 1994, 6, 827 CrossRef CAS.
- D. Shaw, Introduction to Colloid and Surface Chemistry, 3rd edn, Butterworth & Co. Ltd., London, 1980 Search PubMed.
- J. S. Kasper and S. M. Richards, Acta Crystallogr., 1964, 17, 752 CrossRef CAS.
- L.-G. Liu, W. A. Bassett and J. Sharry, J. Geophys. Res., 1978, 83, 2301 CrossRef CAS.
- G. S. Smith and P. B. Isaacs, Acta Crystallogr., 1964, 17, 842 CrossRef CAS.
- Y. Oh, S. Bag, C. D. Malliakas and M. G. Kanatzidis, Chem. Mater., 2011, 23, 2447 CrossRef.
- S. Bag and M. G. Kanatzidis, J. Am. Chem. Soc., 2010, 132, 14951 CrossRef CAS.
- (a) J. Heo and J. D. Mackenzie, J. Non-Cryst. Solids, 1989, 111, 29 CrossRef CAS; (b) Y. Wang, J. Wells, D. G. Georgiev, P. Boolchand, K. Jackson and M. Micoulaut, Phys. Rev. Lett., 2001, 87, 185503–1 CrossRef; (c) V. Krasteva, D. Machewirth and G. H. Sigel, J. Non-Cryst. Solids, 1997, 213–214, 304 CrossRef; (d) V. Krasteva, D. Hensley and G. H. Sigel, J. Non-Cryst. Solids, 1997, 222, 235 CrossRef CAS; (e) A. B. Seddon and M. A. Hemingway, J. Therm. Anal., 1991, 37, 2189 CrossRef CAS; (f) A. B. Seddon and M. A. Hemingway, J. Non-Cryst. Solids, 1993, 161, 323 CrossRef CAS; (g) J. Heo, J. S. Sanghera, H. Nasu and J. D. Mackenzie, Mater. Sci. Forum, 1987, 19–20, 55 CrossRef CAS; (h) F. C. Lin and S.-M. Ho, J. Am. Ceram. Soc., 1963, 46, 24 CrossRef CAS; (i) S. Maneglier-Lacordaire, J. Rivet and J. Flahaut, Ann. Chim. (Cachan, Fr.), 1975, 10, 291 Search PubMed.
- Standard Test Methods for Determining Chemical Durability of Nuclear, Hazardous, and Mixed Waste Glasses and Multiphase Glass Ceramics: The Product Consistency Test (PCT), A. International, C 1285-02, ASTM International, West Conshohocken, PA, 2008, http://www.astm.org Search PubMed.
- M. A. Popescu, Non-Crystalline Chalcogenides (Solid-State Science and Technology Library), Kluwer Academic Publishers, New York, NY, 2000, vol. 8 Search PubMed.
- B. J. Riley, S. K. Sundaram, B. R. Johnson and L. V. Saraf, J. Non-Cryst. Solids, 2008, 354, 813 CrossRef CAS.
- D. Turianitsa, V. V. Himinets and T. V. Timko, J. Appl. Chem. USSR (Engl. Trans.), 1975, 48, 1360 Search PubMed.
- V. V. Himinets and L. B. Baranova, J. Prikladnoi Himii, 1983, 56, 1514 Search PubMed.
- A. Hruby, Czech. J. Phys., 1972, 22, 1187 CrossRef CAS.
- J. S. Wang, E. M. Vogel and E. Snitzer, Opt. Mater., 1994, 3, 187 CrossRef CAS.
- (a) C. Jijian, C. Wei and Y. Dapeng, J. Non-Cryst. Solids, 1995, 184, 124 CrossRef; (b) W. Chen, G. R. Chen and J. J. Cheng, Phys. Chem. Glasses, 1997, 38, 156 CAS; (c) B. Monchev, T. Petkova, P. Petkov and S. Vassilev, J. Mater. Sci., 2007, 42, 9836 CrossRef CAS; (d) V. Boev, M. Mitkova, E. Lefterova, T. Wagner, S. Kasap and M. Vlcek, J. Non-Cryst. Solids, 2000, 266–269, 867 CrossRef CAS; (e) V. S. Vassilev, S. V. Boycheva, Z. G. Ivanova, S. A. Stefanova and I. N. Markova-Deneva, in Proc. of the 12th International Conference on Glass and Ceramics, ed. B. Samuneva, Sci. Invest. Publ. House, Sofia, 1997, pp. 173–176 Search PubMed.
| This journal is © The Royal Society of Chemistry 2011 |