Sol–gel preparation, microstructure and luminescence of rare earth/silica/polyacrylamide hybrids through double functionalized covalent Si–O linkage†
Bing
Yan
*,
Li-Min
Zhao
,
Xiao-Long
Wang
and
Yan
Zhao
Department of Chemistry, Tongji University, Shanghai 200092, China. E-mail: byan@tongji.edu.cn; Fax: +86-21-65982287; Tel: +86-21-65984663
First published on 12th September 2011
Abstract
A new type of photoactive macromolecule hybrid materials have been synthesized with covalent bond linking organically modified Si–O and polymer chains through sol–gel technology. The photoactive center consists of a rare earth (Eu,Tb)-functional silane precursor HIPASi of which 5-hydroxyisophthalic acid (HIPA) is grafted by 3-(triethoxysilyl)-propyl isocyanate (TEPIC). An organic polymer chain (PAMSi), from the polymerization of the precursor AM–Si originated from acrylamide monomer (AM) modified by(3-chloropropyl)trimethoxysilane (CPMS), is introduced with Si–O covalent bond to HIPASi. The resulting hybrids possess a homogeneous and regular microstructure without phase separation phenomena, whose particle sizes decreased with the enhancement of organic polymer component (PAM) content. The luminescence lifetimes, quantum efficiencies and energy transfer efficiencies of the hybrid materials are improved with an increase in the molar ratio of the organic polymer unit, suggesting that the organic polymers are favorable for the luminescence for the template effect of organic chain.
Introduction
Rare earth complexes within various hosts make attractive materials for optical applications owing to their excellent luminescence characteristics from the electronic transitions between the 4f energy levels.1 In recent years, rare earth/polymer hybrid materials or rare earth hybrids with luminescent properties have attracted significant interest because of their potential application in the fluorescence and laser systems.2 Compared with luminescent pure rare earth organic complexes, rare earth ion (or its complex) doped polymers possess an improved processing ability, chemical stability, and mechanical strength for the introduction of polymer.3Research on rare earth/organic polymer hybrids with luminescent properties began in the 1980s.4 In general, there are four methods to prepare rare earth-fabricated polymers:5 blends, polymerization, sol–gel methods, and solution mixing, which can be more simply classified as chemical and physical incorporations. It has been testified that if a rare earth ion is chemically bonded to the chains of a polymer, the content of rare earth ion, the type of chemical interaction between the rare earth ion and polymer chain, and the distribution of rare earth ion along the polymer chains, etc., will strongly influence the luminescent properties of the materials obtained.6 In comparison with chemical incorporation, the physical methods, in particular the direct blending of a rare earth ion organic complex with a polymer, has seldom been investigated, even though this method is convenient and cheap.6 In these kind of rare earth complex/polymer hybrids, each dispersed complex molecule is a luminescent unit, which means that some primary factors can determine the final luminescent behavior of the hybrids, such as the transparency of the hybrids, their dimensions, the amount of dispersed rare earth complex molecule, and the interfacial interaction between the rare earth organic complex and the polymer matrix. With the same content of rare earth complex, a dispersion of smaller-sized particles of the complex leads to a higher transparency of the hybrid and a larger interfacial region between the dispersed complex particles and the matrix polymer, both of which would improve the efficiency of excitation.
On the other hand, some researches are engaged in the inorganic polymeric hybrid materials constructed with rare earth ions and modified silane.7 The rare earth species covalently bonded Si–O network, which can solve the homogeneous dispersion and luminescent stability. Our group also achieved some new technology for the construction of these hybrid materials.8–10 So the tendency for rare earth hybrid materials is how to assemble inorganic polymeric Si–O network and organic polymeric chain into one hybrid systems to compose their functions together effectively.8–10 While these hybrid materials are mainly based on the direct coordination between rare earth ions and the organic polymer chain unit.
Herein, we put forward another path to form the rare earth macromolecular hybrid materials, whose organic chain unit is covalently bonded with inorganic silica through an Si–O network, while not coordinating to rare earth ions. The covalent Si–O bonds between the polymer and the lanthanide complex can have a direct influence on the sol–gel process of organically modified siliane precursors, which is important to form the microstructure of the final hybrids. Generally, the polymer, such as polyacrylamide (PAM), has no effective photoactive group to have energy transfer to the rare earth ions. So mostly, polymer PAM behaves as a structural host unit rather than a functional one. 5-Hydroxyisophthalic acid (HIPA) is modified by 3-(triethoxysilyl)-propyl isocyanate (TEPIC) through the addition reaction between its hydroxyl group and the inner ester group of TEPIC. So the two meta carboxylate groups can produce the chelating effect with rare earth ions and the remaining amide can be left to react with the functional PAM modified by (3-chloropropyl) trimethoxysilane (CPMS) through the cohydrolysis and copolycondensation process. So the two carboxylates and the hydroxyl group are all in the meta position along the whole phenyl cycle, whose space arrangement is of benefit for the design and construction of the aim hybrid system. This is the advantage over other aromatic carboxylic acid such as p-aminobenzoic acid or p-hydroxybenzoic acid, etc.7 The microstructure and especially the luminescent behavior have been studied in detail.
Expeimental
Chemicals and procedures
5-Hydroxyisophthalic acid (HIPA), 3-(triethoxysilyl)-propyl isocyanate (TEPIC), and (3-chloropropyl) trimethoxysilane (CPMS) are supplied by Lancaster Synthesis Ltd. Acrylamide (AM) is purchased from shanghai chemical plant. Other starting reagents are used as received.10 mmol HIPA (1.82 g) is first dissolved in DMF (20 mL) by stirring and 1 mmol TEPIC (2.50 g) is then added to the solution dropwise. The whole mixture is refluxed at 60 °C under nitrogen for 12 h. After filtration to remove the filtrate including HCl, a pale yellow oil product HIPASi (yield: 65%) is obtained and then a stoichiometric amount of Eu(Tb)(NO3)3·6H2O is added to the solution of HIPASi in DMF. The molar ratio of Eu(Tb)(NO3)3·6H2O/HIPASi is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3. The resulting complexes are synthesized with yields of 57% for the Eu complex and 61% for the Tb one, respectively. The elemental analysis, 1H NMR and 13C NMR data of HIPASi are as follows: anal. calc. for C18H27O9NSi: C, 50.34; H, 6.34; N, 3.26. Found: C, 50.47; H, 6.13; N, 3.12%. 1H NMR (DMSO-d6) δ 13.2 (2H,s), 7.97 (1H,s), 7.56 (2H,s), 6.34 (1H,t), 3.26 (2H,m), 2.88 (6H,m), 2.73 (9H,t), 1.47 (2H,m), 0.47 (2H,t). 13C NMR (DMSO): δ 166.4 (C1), 157.6 (C5), 132.2 (C3), 120.8 (C2), 119.9 (C4), 119.3 (C6), 66.5 (CH2 (OEt)), 40.3 (C7), 35.7 (C8), 30.7 (CH3 (OEt)), 14.6 (C9). Anal. calc. for C54H82O29N3Si3Eu: C, 44.02; H, 5.61; N, 2.85. Found: C, 44.13; H, 5.40; N, 2.68%. Anal. calc. for C54H82O29N3Si3Tb: C, 43.81; H, 5.58; N, 2.84. Found: C, 44.06; H, 5.41; N, 2.71%. The formula of Eu(Tb) complex precursor (Eu(Tb)–HIPASi) can be predicted as the formulae of Eu(Tb)(HIPASi)(H2O)2 from the elemental data.
:3. The resulting complexes are synthesized with yields of 57% for the Eu complex and 61% for the Tb one, respectively. The elemental analysis, 1H NMR and 13C NMR data of HIPASi are as follows: anal. calc. for C18H27O9NSi: C, 50.34; H, 6.34; N, 3.26. Found: C, 50.47; H, 6.13; N, 3.12%. 1H NMR (DMSO-d6) δ 13.2 (2H,s), 7.97 (1H,s), 7.56 (2H,s), 6.34 (1H,t), 3.26 (2H,m), 2.88 (6H,m), 2.73 (9H,t), 1.47 (2H,m), 0.47 (2H,t). 13C NMR (DMSO): δ 166.4 (C1), 157.6 (C5), 132.2 (C3), 120.8 (C2), 119.9 (C4), 119.3 (C6), 66.5 (CH2 (OEt)), 40.3 (C7), 35.7 (C8), 30.7 (CH3 (OEt)), 14.6 (C9). Anal. calc. for C54H82O29N3Si3Eu: C, 44.02; H, 5.61; N, 2.85. Found: C, 44.13; H, 5.40; N, 2.68%. Anal. calc. for C54H82O29N3Si3Tb: C, 43.81; H, 5.58; N, 2.84. Found: C, 44.06; H, 5.41; N, 2.71%. The formula of Eu(Tb) complex precursor (Eu(Tb)–HIPASi) can be predicted as the formulae of Eu(Tb)(HIPASi)(H2O)2 from the elemental data.
10 mmol AM (0.70 g) is first dissolved in absolute alcohol by stirring and 10 mmol CPMS (2.0 g) is then added to the solution dropwise. The whole mixture is refluxed at 80 °C under nitrogen for 15 h. After isolation, a pale yellow solid AMSi is obtained with a yield of 92.3%. The product (monomer AMSi, 10 mmol/2.45 g) obtained in the above is subsequently mixed with DMF (20 mL) solution of the initiator, benzoyl peroxide (BPO, 0.01 g), to proceed the polymerization reaction under nitrogen purging. The reaction temperature is maintained at 60 °C for about 3 h. The coating liquid is concentrated under room temperature to remove the solvent DMF using a rotary vacuum evaporator, then thermal treatment is performed at 80 °C until the sample solidified. The resulting product is obtained with a yield of 92%. The 1H NMR and 13C NMR date of AMSi are shown as follows: anal. calc. for C9H19O4NSi: C, 46.33; H, 8.21; N, 6.00. Found: C, 46.17; H, 8.04; N, 5.83%. 1H NMR (DMSO-d6) δ 6.8 (1H,t), 6.32 (1H,t), 6.24 (1H,t), 5.71 (1H,t), 3.62 (9H,t), 2.25 (2H,m), 1.25 (2H,m), 0.25 (2H,t). 13C NMR (DMSO): δ 171.5 (C3), 130.4 (C2), 129.1 (C1), 58.2 (CH3 (OCH3)), 47.2 (C4), 26.3 (C5), 18.1 (C6). (Fig. S1†). The number average molecular weight and molecular dispersity of PAMSi are 1.2 × 104 and 2.43 respectively, as determined with gel-permeation chromatography (GPC) calibrated by polystyrene standards.
Sol–gel polymerizations
A sol–gel derived hybrid material is prepared as follows: the synthesized precursor Eu(Tb)–HIPASi complex (0.6 mmol/0.9 g) and PAMSi (7.2 g, 3.6 g) are dissolved in ethanol, and TEOS (tetraethylorthosilane), H2O are added while stirring, then one drop of diluted hydrochloric acid is added to promote hydrolysis. After hydrolysis, 2 mL DMF and an appropriate amount of hexamethylene-tetramine are added to adjust the pH value to about 6.5. The mixture is stirred to achieve a single phase and thermal treatment is performed at 60 °C until the sample solidified. The hybrid materials prepared from the Eu(Tb)–HIPASi complex are denoted as hybrid HIPA. The hybrid materials, which the molar ratio of PAMSi/HIPASi is 1:2, are denoted as hybrid 2HIPA–Si–1PAM and the hybrid materials, which the mole ratio of PAM/HIPA is 1:1, are denoted as hybrid 1HIPA–Si–1PAM. Besides, the pure polymeric hybrids without Eu3+ (Tb3+) have also been prepared through the sol–gel process between HIPASi (and TEOS) and PAMSi (named as HIPA–Si, 2HIPA–Si–1PAM, 1HIPA–Si–1PAM). The concentration of RE3+ ions in the covalent hybrids are measured under the traditional complexometric titration method. The final hybrid samples are dissolved in nitric acid, then titrated with EDTA solution, using a buffer (pH 5.8) and xylenol-orange as indicator. The contents of RE3+ (RE = Eu, Tb) are determined as follows: 11.04% (Eu–HIPA–Si), 11.51% (Tb–HIPA–Si); 1.08% (Eu–1HIPA–Si–1PAM), 1.22% (Tb–1HIPA–Si–1PAM); 1.43% (Eu–2HIPA–Si–PAM), 1.58% (Tb–2HIPA–Si–PAM).
Physical measurements
Elemental analyses (C, H, N) are carried out by the Elementar Cario EL elemental analyzer. Infrared spectroscopy are obtained in KBr pellets and recorded on a Nexus 912 AO446 FT-IR spectrophotometer in the range of 4000–400 cm−1. 1H NMR spectra are recorded in DMSO-d6 on a Bruker AVANCE-500 spectrometer with tetramethylsilane (TMS) as internal reference. Phosphorescence spectra (5 × 10−4 mol L−1CHCl3 solution) and luminescent excitation and emission spectra are obtained on a Perkin-Elmer LS-55 spectrophotometer: excitation slit width = 10 nm, emission slit width = 2.5 nm. All measurements are completed under room temperature except for phosphorescence spectra that are measured at 77 K. Luminescent lifetimes for hybrid materials are obtained with an Edinburgh Instruments FLS 920 phosphorimeter using a 450 W xenon lamp as an excitation source (pulse width, 3 μs). The quantum yield of pure covalently bonded hybrid materials without Eu3+(Tb3+) (HIPA–Si, 2HIPA–Si–1PAM, 1HIPA–Si–1PAM) and the corresponding Eu3+(Tb3+) hybrid materials are determined using an integrating sphere (150 mm diameter, BaSO4 coating) of Edinburgh Instruments. The quantum yield can be defined as the integrated intensity of the luminescence signal divided by the integrated intensity of the absorption signal. Thermogravimetry (TG) and differential scanning calorimetry (DSC) are obtained on Netzsch, model STA 409C in the following conditions: atmosphere of static air heating/cooling rate of 5 °C min−1, 250 mg of powder crucibles of Al2O3. The microstructure is estimated by scanning electron microscopy (SEM, Philips XL-30, acceleration voltage = 20.0 kV).Results and discussion
The formation process of the hybrids
The formation scheme of the hybrid materials can be illustrated in Fig. 1. Firstly, HIPA is modified by TEPIC and further coordinates with the terbium ion to afford the functional inorganic precursor complex Eu(Tb)–HIPASi (Fig. 1(B)). At the same time, AM is modified by TEPIC and then polymerizes with BPO as the initiator to form functional organic precursors PAMSi (Fig. 1(C)). Because of the coexistence of the terminal silanol groups of Eu(Tb)–HIPASi and PAMSi, the hybridization/ polycondensation reactions occur when de-ionized water is added into the mixed system of Eu(Tb)–HIPASi and PAMSi, whose reaction process may be illustrated in Fig. 1(D). Through this method, the rare earth complex and polymer can be connected by the Si–O bonds of the inorganic matrix. Besides, the coordination between HIPASi unit and Eu3+ (Tb3+) ions depends on the two carboxylates from HIPA groups for they possess stronger coordination ability than the amide group although they locate at the similar meta position along the phenyl cycle. The chelation of the two carboxylates to Eu3+ (Tb3+) ions make the whole complex system stable, which is the same as the simple double aromatic carboxylic acid ligands such phthalic acid. So it is not hard to understand the scheme for the hybrid materials and this can be verified by FITR spectra.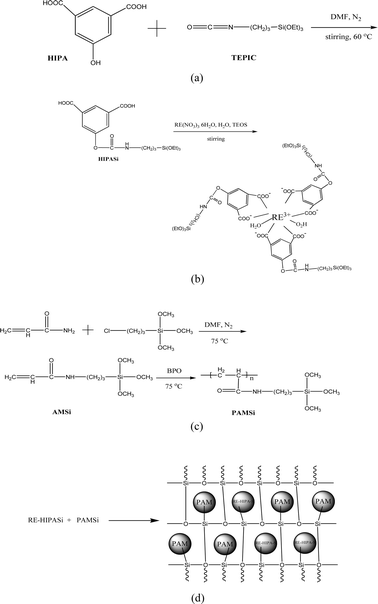 | ||
| Fig. 1 The scheme of the synthesis process of precursors HIPASi (a), RE–HIPASi (b), PAMSi (c) and the final hybrids RE–1HIPA–Si–1PAM (d). | ||
The FT-IR spectra for the precursors and the hybrids
The Fourier-transform infrared (FTIR) spectra of the HIPA and HIPASi are shown in the Fig. 2(a). As for the HIPA spectrum, peaks located at 3440 cm−1 belong to the stretching vibration of OH (νOH) and the bands located at 1700 cm−1, 1660 cm−1, 1590 cm−1 are assigned to the stretching vibration of carbonyl groups (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and the absorption of carboxyl, respectively. In contrast to the spectrum of HIPA, the HIPA–Si spectrum has a strong and broad band at about 1108 cm−1, which is due to the emergence of the Si–O band in the HIPASi. The emergence of the broad band located at 3337 cm−1 in the HIPASi is due to the stretching vibration of NH (νNH) and this means that Si has been successfully grafted onto HIPA. The chemical shifts of the 13C NMR and 1H NMR spectra also testify the structure of HIPASi (Fig. S1†). Fig. 2(b) illustrates the FTIR spectra of AM, AMSi and PAMSi. Upon comparing the AMSi spectrum with the spectrum of AM, the absorbance band located at about 3395 cm−1 has been changed from double peaks to a broad peak, which corresponds to the transformation from –NH2– of AM to –NH– of AMSi. As for the PAM spectrum, disappearance of the peaks located at 1616 cm−1, which belong to the stretching vibration of CC (νCC), and emergence of a strong bond located at 1084 cm−1, which belongs to the absorbance of Si–O, means that polymerization has occurred. Coordination of the rare earth ions by the ligands is clearly shown by infrared spectroscopy. In the spectrum of precursors (b), the νas(COO−) vibration is located at 1686 cm−1 and the νs(COO−) vibration at 1440 cm−1.
O) and the absorption of carboxyl, respectively. In contrast to the spectrum of HIPA, the HIPA–Si spectrum has a strong and broad band at about 1108 cm−1, which is due to the emergence of the Si–O band in the HIPASi. The emergence of the broad band located at 3337 cm−1 in the HIPASi is due to the stretching vibration of NH (νNH) and this means that Si has been successfully grafted onto HIPA. The chemical shifts of the 13C NMR and 1H NMR spectra also testify the structure of HIPASi (Fig. S1†). Fig. 2(b) illustrates the FTIR spectra of AM, AMSi and PAMSi. Upon comparing the AMSi spectrum with the spectrum of AM, the absorbance band located at about 3395 cm−1 has been changed from double peaks to a broad peak, which corresponds to the transformation from –NH2– of AM to –NH– of AMSi. As for the PAM spectrum, disappearance of the peaks located at 1616 cm−1, which belong to the stretching vibration of CC (νCC), and emergence of a strong bond located at 1084 cm−1, which belongs to the absorbance of Si–O, means that polymerization has occurred. Coordination of the rare earth ions by the ligands is clearly shown by infrared spectroscopy. In the spectrum of precursors (b), the νas(COO−) vibration is located at 1686 cm−1 and the νs(COO−) vibration at 1440 cm−1.
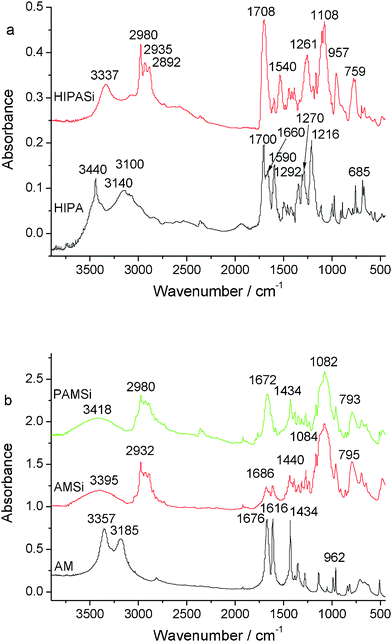 | ||
| Fig. 2 The FTIR spectra of the precursors HIPA, HIPASi (a) and AM, AMSi, PAMSi (b). | ||
Fig. 3 shows the FT-IR spectra of the four rare earth hybrids, which show similar features. The broad absorptions of the ν(Si–C) vibration are located in the range of 1200–1190 cm−1, suggesting that the ν(Si–C) absorption is consistent with the fact that no (Si–C) bond cleavage occurs. The ν(O–H) vibration around 3350 cm−1 indicates the existence of residual silanol groups and the presence of H2O molecule, and the ν(Si–OH) stretching vibration at 960 cm−1 is further evidence of the incompleteness of the condensation reactions. Furthermore, the complexation of RE3+ with the organic compound in all of the hybrids can also be shown by infrared spectroscopy. The νas(COO−) and the νs(COO−) located at 1592 and 1388 cm−1 become much stronger, which can be ascribed to the complexation of the RE3+ ion with the oxygen atom of the COO−group in the hybrids. The Ar–O–CH2 vibrations in all of the hybrids are hardly shifted, revealing that the oxygen atom of the Ar–O–CH2 group is not coordinated to the RE3+ ion. As we know, it is very difficult to prove the exact structure of these kind of non-crystalline hybrid materials, and it is hardly possible to solve the coordination behavior of rare earth ions. However, the main composition and coordination effect of them according to the rare earth coordination chemistry principle and the functional groups of the organic unit can be predicted (see Fig. 1). However, the presence of residual Si–OH groups in the complexes cannot be excluded.
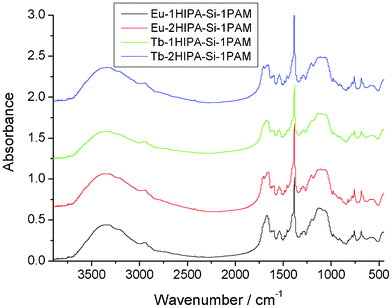 | ||
| Fig. 3 The FT-IR spectra of the four rare earth hybrid materials. | ||
The TG-DSC curves of the hybrids
Fig. S2 shows the thermogravimetry trace (TG) and differential scanning calorimetric (DSC) plot of (A) Eu–HIPA–Si and (B) Eu–HIPA–Si–PAM.† The TG data ofthe Eu–HIPA–Si shows an obvious mass loss of 26.87% in the range of 120–380 °C corresponding to the water molecules and one HIPASi molecule coordinated with Eu3+ (theoretic weight of 24.76% according to the molecular weight). The TG curve of Eu–HIPA–Si–PAM shows two obvious mass losses: around 250 °C and 500 °C. The first weight change in the range of 120–380 °C with a mass loss of 37.85% is attributed to the loss of water molecules and two HIPASi molecules, and the second event in the range of 400–570 °C with a mass of 17.27% corresponds to the thermal decomposition of the organic composition involving the polymer PAMSi. Moreover, the hybrids demonstrated a residual mass of 43.23% and 41.83% when the temperature climbed to 600 °C, indicating that the addition of the polymer PAM does not decrease the thermodynamic character of the hybrid, although the organic polymer has less thermodynamic stability than the inorganic material. The energy changes indicated by the DSC curve also confirmed the results of TG. Furthermore, as can be seen from the supplementary information Fig. S3,† the TG curve of the complex Eu–HIPA has shown two obvious mass losses: in the range of 80–350 °C with a mass loss of 31.23% attributed to the loss of water molecules and HIPASi molecules, and in the range of 350–600 °C with a mass of 43.75% corresponding to the thermal decomposition of the whole configuration of the hybrid. The complex Eu–HIPA exhibited a residue of 22.87% when the temperature climbed to 600 °C. Therefore, the experimental results indicate that the thermodynamic character and stability of the hybrid material has been improved by bonding the polymer PAM into the inorganic Si–O network.The scanning electron microscope (SEM) images of the hybrids
The scanning electron microscope (SEM) images in Fig. 4 show the microstructure of (1) Eu–HIPA–Si hybrid materials, (2) Eu–2HIPA–Si–1PAM hybrid materials where the molar ratio of PAM/HIPA is 1:2 and (3) Eu–1HIPA–Si–1PAM hybrid materials where the mole ratio of PAMSi/HIPASi is 1:1. It can be seen from Fig. 4(1) that the HIPASi hybrid material has a relatively regular trunk or stripe. The precursor HIPA–Si is a derivative from HIPA and its corresponding europium complex generally forms a polymeric structure,11 and this tendency will compete with the construction of a polymeric network structure of Si–O–Si in the hydrolysis and copolycondensation processes of Eu–HIPA–Si. Finally, the hybrid material Eu–HIPA–Si retains the intensive tendency of growing into coordination polymer and reserves the coordinated positions in corresponding bulk materials observed from the microstructure view. Comparing with HIPA hybrid material, when PAMSi is introduced into the Eu–HIPA–Si hybrid material through a hydrolysis/polycondensation process, it can be found that the emergence of circular particles, and the number of particles is increased and the particle size decreases with the enhancement of PAMSi content. The organic polymer chain unit PAMSi possesses two roles, one is the polymer chain to have the ability of orientation, the other is the silanol group of PAMSi to allow copolymerization with HIPASi. Subsequently, the double control of organic polymer chain and copolymerization ability influences the growth and formation of particles in the hybrid systems. As shown in Fig. 4(3), the particle sizes belong to the nanometer dimension and the microstructure has the tendency to be more regular and homogenous. While this kind of ordered micromorphology is hard to observe in a pure organic polymer matrix.
 | ||
| Fig. 4 Selected SEM images of (1) Eu–HIPA–Si hybrid materials, (2) Eu–2HIPA–Si–1PAM hybrid materials with the molar ratio of PAM/HIPA is 1:2 and (3) Eu–1HIPA–Si–1PAM hybrid materials with the molar ratio of PAMSi/HIPASi is 1:1. | ||
It needs to be mentioned that such inorganic/organic polymeric hybrids covalently linked by an Si–O network between an inorganic host and an organic polymer chain have rarely been reported. The detailed cohydrolysis and copolycondensation process can be further described in the following. As mentioned in the experimental, the hybrid materials could be achieved through a copolycondensation reaction between the terminal silanol groups of RE–HIPASi and PAMSi. At the beginning of the reaction, the individual hydrolysis of HIPASi (and TEOS) and PAMSi are predominant. The step II for RE–HIPASi, is related to the copolycondensation reactions between the hydroxyl groups of both RE–HIPASi and TEOS. The covalently bonded hybrids RE–HIPASi bearing the RE–O coordination bond and Si–O covalent bonds can also been obtained after the introduction of RE3+. The step II for RE–HIPA–Si–PAM, is related to the copolycondensation reactions between the hydroxyl groups of both RE–HIPASi (and TEOS) and PAMSi. The covalently bonded hybrids RE–HIPA–Si–PAM with the RE–O coordination bond and Si–O covalent bonds can also been obtained after the introduction of RE3+. Here we named the cooperation of both RE–HIPA–Si–PAM and PMASi (and TEOS) within the in situ sol–gel process as cohydrolysis and copolycondensation (similar to copolymerization of organic monomer). Besides, the coordination reaction between RE3+ and HIPASi exists in the in situ copolycondensation process, which has an influence on the sol–gel process and the microstructure or physical properties of the hybrids. In particular it is worthy pointing out that the organic polymer chain (PAM) is introduced in the hybrid system through the copolycondensation reaction with inorganic polymeric host by Si–O covalent bonds and that a coordination reaction between RE3+ and the PAM chain does not exist. Compared to the RE–HIPA–Si without organic chain and RE–2HIPA–Si–1PAM hybrids, an apparent distinction exists. The former only involves the sol–gel process (hydrolysis and polycondensation) of inorganic precursors HIPASi and TEOS to form the inorganic polymeric Si–O host, while the latter appears during the cohydrolysis and copolymerization reaction between inorganic precursors (HIPASi and TEOS) and PAMSi, which have a great influence on the sol–gel process and the microstructure or physical properties of the hybrids. Because the long organic polymer chain grafted on the HIPASi precursor achieves the long tail of HIPA–Si–PAM and will fulfil the template effect to affect the sol–gel process. Subsequently, the interpenetrating polymeric network microstructure may easily form and the microstructure of the resulting hybrids tends to regular. Besides, the RE–1HIPA–Si–1PAM hybrids will be expected to be more regular than RE–1HIPA–Si–1PAM with the increase of organic chain as the template effect of the PAM chain is enhanced. The interpretation has been verified from the above SEM patterns and the below luminescent properties. The Si–O connects the RE complex (RE–HIPA–Si) and the polymer unit (PAM) and the regular microstructure of the final hybrids are easily realized.
The phosphorescence spectra of the precursor and linkages
Fig. S3 shows the low temperature phosphorescence spectra of (A) HIPA and (B) HIPASi at 77 K.† Molecular phosphorescence belongs to the character of the organic molecular ligands and different phosphorescence bands correspond to different ligand molecules, so a blue shift of 9 nm appears between A (434 nm) and B (425 nm) in terms of modification from HIPA to HIPASi. According to intramolecular energy transfer mechanism,11 the corresponding intramolecular transfer efficiency from the HIPASi to Tb3+ mainly depends on the energy match between the triplet state energy of HIPASi (corresponded to the phosphorescence band, 23580 cm−1) and the resonant emissive energy level of the central Eu3+ (17167 cm−1), Tb3+ (20500 cm−1). Therefore, it can be predicted that HIPASi shows a good energy couple and can sensitize the luminescence of Eu3+ (Tb3+) effectively.12The luminescence spectra of the hybrids
The selected excitation spectra of Eu–HIPA–Si, Eu–2HIPA–Si–1PAM and Eu–1HIPA–Si–1PAM hybrids are shown in Fig. 5. All these excitations are obtained by monitoring the maximum emission of the Eu3+ ions at 614 nm and a dominant broad band appears from 250 to 400 nm except for the distinction of maximum excitation peaks, which can be due to the absorption of photoactive HIPA unit in the organic modified Si–O network. The strong absorption of photoactive HIPA unit behaves as the main energy absorption for the whole hybrid systems. With the content of organic polymer unit PAMSi, the maximum excitation shows the red shift to longer ultraviolet region, which suggests that the absorption of organic modified Si–O network by the PAM chain tends to the more stable energy state suitable for the energy transfer within the whole hybrid systems. Undoubtedly, the red shift to low energy state is favorable for the luminescence of Eu3+. The excitation spectra for the three covalently bonded terbium hybrid materials show the similar feature. | ||
| Fig. 5 The excitation spectra of Eu–HIPA–Si hybrids, Eu–2HIPA–Si–1PAM hybrids and Eu–1HIPA–Si–1PAM hybrids. | ||
Fig. 6 show the selected emission spectra of the hybrid material RE–1HIPA–Si–1PAM with the molar ratio of PAM–Si:HIPA–Si is 1:1 compared with the spectra of the rare earth hybrids without PAM chain. The emission spectra of the hybrid material RE–1HIPA–Si–1PAM and the rare earth hybrids without PAM chain Eu–HIPA–Si also show the characteristic emission peaks located at 588, 614, 717 nm in (b) corresponding to the 5D0 → 7FJ (J = 1, 2, 4) transitions. Among these peaks, the red emission at 616 nm due to the 5D0 → 7F2 electric dipole transition is with the highest relative intensities, higher than the orange emission of magnetic dipole transition of 5D0 → 7F1 at about 589 nm, which has indicated Eu3+ site in an environment with low inversion symmetry. Generally speaking, the forbidden 5D0 → 7F2 electric dipole transition is rather sensitive to the coordinative environment of the Eu ions, so it is apparent that the polymer–inorganic based asymmetric micro-chemical environment leads to the polarization of the Eu ion, which has enhanced the probability for the 5D0 → 7F2 transition. The emission spectra of hybrid material Tb–1HIPA–Si–1PAM and the rare earth hybrids without PAM chain Tb–HIPA–Si show the characteristic emission peaks located at 488, 543, 584, 618 nm in (a) corresponding to the 5D4 → 7FJ (J = 6, 5, 4, 3) transitions. Among these emission peaks, the green emission (5D4 → 7F5) with the highest relative intensities has been observed, which has indicated that the effective energy transfer took place between the HIPASi and the chelated Tb3+.
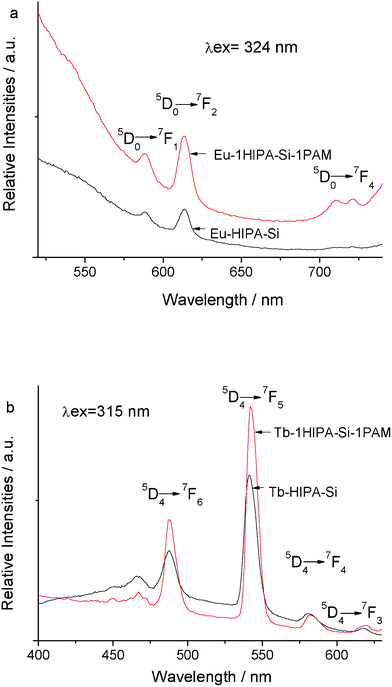 | ||
| Fig. 6 Selective emission spectra of Eu–1HIPA–Si–1PAM hybrids and pure europium hybrids without PAM chain Eu–HIPA–Si (a) and Tb–1HIPA–Si–1PAM hybrids and pure terbium hybrids without PAM chain Tb–HIPA–Si (b). | ||
Moreover, from the emission spectra, it is found that the relative intensities of hybrid material RE–1HIPA–Si–1PAM are higher than those of the rare earth hybrid without polymer chain RE–HIPA–Si. The factor could be the rigid structure of silica gel which has been formed between HIPASi, PAMSi and TEOS, has limited the vibration of the hydroxyl group around Eu3+ or Tb3+, replaced the water molecule in the material and prohibited non-radiative transitions. Accordingly, we may expect that through this efficient way, leaching of the photoactive molecules or clustering of the emitting centers could be avoided, and high concentration of metal ions could be realized. Besides, the triplet state energy of HIPASi is 23580 cm−1 form the phosphorescence data and the resonant emissive energy level of the Eu3+ is 17300 cm−1, 20500 cm−1 for Tb3+, and the energy difference between the modified ligand HIPASi and Eu3+ is 6280 cm−1, between the modified ligand HIPASi and Tb3+ with 3080 cm−1. It is apparent that the energy difference between the modified ligand HIPASi and Eu3+ is a little larger for the energy transfer and the energy difference between the modified ligand HIPASi and Tb3+ is more appropriate, resulting in a more effective intramolecular energy transfer mechanism in the hybrid material containing Tb3+ than that in the hybrid material containing Eu3+. Therefore, the fluorescent properties including the peak shape, the relative intensities and the baseline height of hybrid material Tb–1HIPA–Si–1PAM are better than those of the hybrid material Eu–1HIPA–Si–1PAM observed from the emission spectra.
The luminescent lifetimes and quantum efficiencies
The typical decay curve of the Eu and Tb hybrid materials are measured and they can be described as a single exponential (ln(S(t)/S0) = −k1t = −t/τ), indicating that all Eu3+ and Tb3+ ions occupy the same average coordination environment. The resulting lifetime data of Eu and hybrids are given in Table 1. It is found that the lifetimes of hybrids Eu–1HIPA–Si–1PAM materials are longer than those of Eu–2HIPA–Si–1PAM and Eu–HIPA–Si hybrids, suggesting that the introduction of an organic polymeric chain can enhance the luminescence stability of the whole hybrid system. Furthermore, we selectively determined the emission quantum efficiencies of the 5D0 excited state of the europium ion for Eu3+ hybrids on the basis of the emission spectra and lifetimes of the 5D0 emitting level, the detailed luminescent data are shown in Table 1. The quantum efficiency of the luminescence step, η expresses how well the radiative processes compete with non-radiative processes.13 Non-radiative processes influence the experimental luminescence lifetime and so the quantum efficiency can be calculated from the radiative transition rate constant and experimental luminescence lifetime.13,14 The branching ratio for the 5D0 → 7F5,6 transitions can be neglected as both are not detected experimentally, whose influence can be ignored in the depopulation of the 5D0 excited state.13 Since 5D0 → 7F1 belongs to the isolated magnetic dipole transition, it is practically independent of the chemical environments around the Eu3+ ion, and thus can be considered as an internal reference for the whole spectrum, the experimental coefficients of spontaneous emission can be calculated according to the relation. 14 With the content of organic polymer unit PAM, the luminescent quantum efficiency enhances, indicating that the organic chain may be beneficial for the luminescence of Eu3+ species. Furthermore, we determined the Judd–Ofelt parameters for the three covalently bonded europium hybrid materials. The Ω2, Ω4 intensity parameters for the three hybrid materials are shown in Table 1. The distinction of the two intensity parameters for the three hybrids is not apparent, suggesting that the Eu3+ ion is located in a polarizable chemical environment for luminescence.| Hybrids | Eu–HIPA–Si | Eu–2HIPA–Si–1PAM | Eu–1HIPA–Si–1PAM |
|---|---|---|---|
| a For 5D0 → 7F2 transition of Eu3+. b () Calculated value for luminescent efficiency. | |||
| τ (ms)a | 0.27 | 0.33 | 0.36 |
| A rad (s−1) | 218 | 229 | 242 |
| τ exp −1(s−1) | 3704 | 3030 | 2778 |
| A nrad (s−1) | 3486 | 2801 | 2536 |
| η (%) | 4.3 (5.9)b | 5.8 (7.5)b | 6.8 (8.7)b |
| Ω 2 | 13.1 | 13.9 | 14.6 |
| Ω 4 | 0.48 | 0.56 | 0.73 |
| η ET (%) | 21.2 | 23.6 | 24.8 |
In order to show the influence of microstructure on the luminescent behavior, we selectively determined the energy transfer efficiency for the three europium centered covalently bonded hybrid materials. For all the covalently bonded hybrids, the energy acceptor are the same Eu3+ (Tb3+) and energy donor (or host) is different, HIPA–Si, 2HIPA–Si–1PAM and 1HIPA–Si–1PAM, respectively. On the basis of the references 15,
| ηET = ηDA/ηD |
Conclusions
We have developed a feasible method for the construction of luminescent rare earth macromolecular hybrid materials with strong chemical bonds through cohydrolysis and copolycondensation reactions using a sol–gel process. Among the inorganic precursor unit HIPASi is the grafting derivatives of HIPA by TEPIC and the organic precursor unit from the modified functional monomer AMSi of acrylamide monomer by CPMS to form the amidation derivative. The microstructure and photophysical properties of the resulting hybrids are compared to different compositions of polymeric components. With the introduction of organic polymeric chain PAMSi, the microstructure becomes regular and the particle size decreases to nanometer dimensions. Especially the luminescent lifetimes, quantum efficiencies and energy transfer efficiencies are improved with the molar ratio of PAMSi.Acknowledgements
This work is supported by the National Natural Science Foundation of China (20971100, 20671072, 20301013) and Program for New Century Excellent Talents in University (NCET-08-0398).References
- E. H. Huffman, Nature, 1963, 200, 158 CAS.
- (a) M. X. Yu, F. Y. Li, Z. G. Chen, H. Hu, C. Zhan, H. Yang and C. H. Huang, Anal. Chem., 2009, 81, 930 CrossRef CAS; (b) J. C. Wu, Q. W. Tian, H. Hu, Q. Xia, Y. Zou, F. Y. Li, T. Yi and C. H. Huang, Chem. Commun., 2009, 4100 RSC; (c) M. R. Robinson, J. C. Ostrowski, G. C. Bazan and M. D. McGehee, Adv. Mater., 2003, 15, 1547 CrossRef CAS.
- (a) M. R. Robinson, M. B. O’Regan and J. C. Bazan, Chem. Commun., 2000, 1645 RSC; (b) X. F. Qiao and B. Yan, Dalton Trans., 2009,(40), 8509 RSC.
- (a) Y. Ueba, E. Banks and Y. Okmoto, J. Appl. Polym. Sci., 1980, 25, 2007 CrossRef CAS; (b) Y. Okmoto, Y. Ueba, N. F. D. Zhanibekov and E. Banks, Macromolecules, 1981, 14, 17 CrossRef CAS.
- (a) J. M. Huang, V. Bekeari, P. Lianos and S. Carios, J. Lumin., 1999, 81, 285 CrossRef CAS; (b) R. Moleski, E. Stathatos, V. Bekeari and P. Lianos, Thin Solid Films, 2002, 416, 279 CrossRef CAS; (c) A. C. Franville, D. Zambon, R. Mahiou and Y. Troin, Chem. Mater., 2000, 12, 428 CrossRef CAS; (d) H. R. Li, J. Lin, H. J. Zhang, H. C. Li, L. S. Fu and Q. G. Meng, Chem. Commun., 2001, 1212 RSC; (e) V. Bekeari, P. Lianos, U. L. Stangar, B. Orel and P. Judeinstein, Chem. Mater., 2000, 12, 3095 CrossRef CAS; (f) B. Yan and X. F. Qiao, J. Phys. Chem. B, 2007, 111, 12362 CrossRef CAS.
- (a) X. F. Qiao and B. Yan, J. Phys. Chem. B, 2009, 113, 11865 CrossRef CAS; (b) C. Guizard and P. Lacan, New J. Chem., 1994, 18, 1097 Search PubMed; (c) H. Y. Chen and R. D. Archer, Macromolecules, 1996, 29, 1957 CrossRef CAS; (d) V. Bekiari, G. Pistolis and P. Lianos, Chem. Mater., 1999, 11, 3189 CrossRef CAS; (e) L. H. Wang, W. Wang, W. G. Zhang, E. T. Kang and W. Huang, Chem. Mater., 2000, 12, 2212 CrossRef CAS.
- (a) L. D. Carlos, R. A. S. Ferreira, V. D. Bermudez and S. J. L. Ribeiro, Adv. Mater., 2009, 21, 509 CrossRef CAS; (b) K. Binnemans, Chem. Rev., 2009, 109, 4283 CrossRef CAS.
- (a) Q. M. Wang and B. Yan, J. Mater. Chem., 2004, 14, 2450 RSC; (b) Q. M. Wang and B. Yan, Cryst. Growth Des., 2005, 5, 497 CrossRef CAS; (c) B. Yan, Q. M. Wang and D.J. Ma, Inorg. Chem., 2009, 48, 36 CrossRef CAS; (d) J. L. Liu and B. Yan, J. Phys. Chem. B, 2008, 112, 10898 CrossRef CAS.
- (a) B. Yan and H. F. Lu, Inorg. Chem., 2008, 47, 5601 CrossRef CAS; (b) J. L. Liu and B. Yan, J. Phys. Chem. C, 2008, 112, 14168 CrossRef CAS; (c) B. Yan and Q. M. Wang, Cryst. Growth Des., 2008, 8, 1484 CrossRef CAS; (d) Y. Li, B. Yan and H. Yang, J. Phys. Chem. C, 2008, 112, 3959 CrossRef CAS.
- (a) Y. J. Li and B. Yan, Inorg. Chem., 2009, 48, 8276 CrossRef CAS; (b) B. Yan and Q. M. Wang, J. Photochem. Photobiol., A, 2008, 197, 213 CrossRef CAS; (c) B. Yan, L. M. Zhao and J. L. Liu, J. Photochem. Photobiol., A, 2008, 199, 50 CrossRef CAS; (d) X. F. Qiao and B. Yan, Eur. Polym. J., 2009, 45, 2002 CrossRef CAS.
- (a) X. F. Qiao and B. Yan, J. Phys. Chem. B, 2008, 112, 14742 CrossRef CAS; (b) X. F. Qiao and B. Yan, Inorg. Chem., 2009, 48, 4714 CrossRef CAS.
- (a) J. W. Moore, M. D. Glick and W. A. Baker, J. Am. Chem. Soc., 1972, 94, 1858 CrossRef CAS; (b) J. Kay, J. W. Moore and M. D. Glick, Inorg. Chem., 1972, 11, 2818 CrossRef CAS; (c) S. Sato and W. Mada, Bull. Chem. Soc. Jpn., 1970, 43, 2403; (d) W. M. Faustino, O. L. Malta and G. F. de Sa, J. Chem. Phys., 2005, 122, 050409 Search PubMed; (e) D. J. Dexter, J. Chem. Phys., 1953, 21, 836 CrossRef CAS; (f) C. R. S. Dean and T. M. Shepherd, J. Chem. Soc., Faraday Trans. 2, 1975, 71, 146 RSC.
- (a) M. H. V. Werts, R. T. F. Jukes and J. W. Verhoeven, Phys. Chem. Chem. Phys., 2002, 4, 1542 RSC; (b) E. E. S. Teotonio, J. G. P. Espynola, H. F. Brito, O. L. Malta, S. F. Oliveria, D. L. A. de Foria and C. M. S. Izumi, Polyhedron, 2002, 21, 1837 CrossRef CAS.
- (a) O.L. Malta, H.F. Brito, J.F.S. Menezes, F.R. Goncalves e Silva, S. Alves, F.S. Farias and A.V.M. Andrade, J. Lumin., 1997, 75, 255 CrossRef CAS; (b) G. F. de Sa, O. L. Malta, C. D. Donega, A. M. Simas, R. L. Longo, P. A. Santa-Cruz and E. F. da Silva, Coord. Chem. Rev., 2000, 196, 165 CrossRef CAS; (c) J.C. Boyer, F. Vetrone, J.A. Capobianco, A. Speghini and M. Bettinelli, J. Phys. Chem. B, 2004, 108, 20137 CrossRef CAS.
- (a) P. R. Biju, G. Jose, V. Thomas, V. P. N. Nampoori and N. V. Unnikrishnan, Opt. Mater., 2004, 24, 671 Search PubMed; (b) P. P. Lima, S.S. Nobre, R.O. Freire, S.A. Junior, L. Mafra, R.A.S. Ferreira, U. Pischel, O. L. Malta and L.D. Carlos, J. Phys. Chem. C, 2007, 111, 17627 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1ra00196e |
| This journal is © The Royal Society of Chemistry 2011 |