Synthesis of ABC type miktoarm star copolymers by triple click chemistry†
Birol
Iskin
,
Gorkem
Yilmaz
and
Yusuf
Yagci
*
Department of Chemistry, Istanbul Technical University, 34469, Maslak, Istanbul, Turkey. E-mail: yusuf@itu.edu.tr; Fax: +90–212–2856386; Tel: +90–212–2853241
First published on 17th October 2011
Abstract
An ABC type miktoarm star copolymer possessing polystyrene (PS), poly(ε-caprolactone) (PCL) and poly(ethylene glycol) (PEG) arms was synthesized by combining existing triple click chemistries, namely thiol–ene, copper catalyzed azide–alkyne cycloaddition (CuAAC) and Diels–Alder (DA) reactions. For this purpose, a core (1-(allyloxy)-3-azidopropan-2-yl (anthracen-9-ylmethyl) succinate) with allyl, azide and anthracene functionalities was synthesized in two steps with high yields. Then, polymers with corresponding clickable sites, namely ω-thiol polystyrene (PS-SH), α-alkyne poly(ε-caprolactone) (alkyne-PCL) and ω-maleimide poly(ethylene glycol) (Me-PEG-MI), were independently prepared. As the first step of the grafting onto process, PS-SH was thiol–ene clicked onto the core to yield PS-N3-Ant. Finally, alkyne-PCL and Me-PEG-MI were bonded to PS-N3-Ant either in a sequential or a one-pot in situ manner using CuAAC and DA click reactions. All intermediates, related polymers at different stages and final PS-PCL-PEG miktoarm star copolymer were characterized by 1H NMR, FT-IR, and GPC analyses.
Introduction
Synthesis of complex macromolecular architectures with narrow molecular weight distributions and well-defined structures has been a key challenge for the last decade.1,2 The discovery of controlled radical polymerization techniques (CRP)3–9 and its combination with high yield reactions introduces a great variety of possibilities to achieve synthesis of such polymeric structures.10–15 Recently, two main processes have been honored as “click” reactions, due to their modularity, versatility and high efficiency under mild reaction conditions. These are Copper Catalyzed Alkyne–Azide Cycloaddition (CuAAC)16,17 and Diels–Alder (DA) reactions,10,18,19 which occur between a conjugated diene and a dienophille. More recently, thiol–ene reactions have also been introduced as an alternative click route that can be performed at moderately low temperatures with and without photoinitiators.20–26 In addition, thiol–ene reactions have high priority especially in biochemistry, since they generally occur in living systems.27–30A number of research papers describe the application of double click reactions for the modification and synthesis of various polymeric structures by combining two of these click processes. Among them, integration of CuAAC and DA is the most common grouping for the synthesis of a variety of macromolecular structures as reflected by numerous publications.31–33 However, the combination of the corresponding thiol–ene reaction with the other click reaction has been scarcely investigated.26,34,35 In a recent paper, we reported the first example of the combination of thiol–ene click chemistry with CuAAC reaction for the synthesis of an ABC type star copolymer.34 In order to further extend the use of thiol–ene chemistry in macromolecular synthesis, the present work focuses on the application of such simple click reaction to a superior level for the synthesis of an ABC type miktoarm star copolymer by combining CuAAC and DA processes.
Experimental section
Materials
Allyl glycidyl ether (99%, Sigma-Aldrich), sodium azide (97%, Sigma-Aldrich), 9-anthracene methanol (97, Aldrich), succinic anhydride (≥99%, Aldrich), ethanolamine (≥99%, Aldrich), potassium xanthate (98%, Fluka), 1,2-ethandithiol (99%, Sigma-Aldrich), bis-(2,4,6-trimethylbenzoyl)-phenylphosphine oxide (Ciba), copper (I) bromide (98%, Acros), N,N,N′,N′′,N′′′-pentamethyldiethylenetriamine (PMDETA, 99%, Aldrich), propargyl alcohol (99%, Aldrich), tin(II) 2-ethylhexanoate (98%, Aldrich), poly(ethylene glycol) methyl ether (Me-PEG, Mn ≈ 550 g mol−1, Aldrich), N,N′-dicyclohexylcarbodiimide (DCC, 99%, Aldrich), 4-dimethylaminopyridine (DMAP, 99%, Aldrich), and methanol (99%, Reiden-de Haën) were used as received. Styrene (99%, Aldrich) was passed through a basic alumina column to remove the inhibitor. ε-Caprolactone (99%, Aldrich) was distilled over calcium hydride prior to use.Characterizations
1H NMR measurements were recorded in CDCl3 with Si(CH3)4 as internal standard, using a Bruker AC250 (250.133 MHz) instrument. FT-IR spectra were recorded on a Perkin-Elmer FT-IR SpectrumOne-B spectrometer. Molecular weights were determined by a gel permeation chromatography (GPC) instrument, a Viscotek GPCmax Autosampler system, consisting of a pump, three ViscoGEL GPC columns (G2000HHR, G3000HHR and G4000HHR), and a Viscotek differential refractive index (RI) detector with a THF flow rate of 1.0 mL min−1 at 30 °C. The RI detector was calibrated with PS standards having narrow molecular weight distribution. Data were analyzed using Viscotek OmniSEC Omni-01 software. Differential scanning calorimetry (DSC) was performed on a Perkin-Elmer Diamond DSC.Synthesis of intermediates and precursor polymers
Synthesis of 4-(anthracen-9-ylmethoxy)-4-oxobutanoic acid (AOB),362-(2-hydroxyethyl)-3a,4,7,7a-tetrahydro-1H-4,7-epoxyisoindole-1,3(2H)-dione (HTEO)37 and 4-(2-(1,3-dioxo-3a,4,7,7a-tetrahydro-1H-4,7-epoxyisoindol-2(3H)-yl)ethoxy)-4-oxobutanoic acid (DEO)10 was carried out as described in the literature.ω-Bromo functional polystyrene (PS-Br)38 (Mn,NMR = 2030, Mn,GPC = 1910 g mol−1, Mw/Mn = 1.19) and α-alkyne functional poly(ε-caprolactone) (alkyne-PCL)39,40Mn,NMR = 4730; Mn,GPC = 6200 (relative to linear PS); Mw/Mn = 1.22 were prepared according to modified literature procedures.
Synthesis of 1-(allyloxy)-3-azidopropan-2-ol (AAP)
Synthesis of 1-(allyloxy)-3-azidopropan-2-ol (AAP) was carried out as described previously.34 Thus, allyl glycidyl ether (5.0 ml, 1 eq.) was dissolved in ethanol (150 mL) in a 500 ml round bottom flask equipped with a magnetic stirrer. Then, sodium azide (12 g, 3 eq.) in water (120 mL) was simply added to the solution via an additional funnel and the reaction mixture was left stirring overnight. The reaction solvent was three times extracted with CH2Cl2 and the organic phase was dried over MgSO4. After filtration, the organic layer was passed through a short column of silica and CH2Cl2 was evaporated under reduced pressure to give a viscous yellow liquid as the product. (Yield: 96%) 1H NMR (CDCl3, 250 MHz): δ = 5.83 (m, 1H, –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ), 5.24 (m, 1H, trans CH2), 5.16 (m, 1H, cis CH2) 3.94 (d, 2H, CH2CH–CH2–O–), 3.40 (p, 1H, –CH–OH), 3.88 (d, 2H, –O–CH2–), 3.30 (d, 2H, –CH2–N3) 3.13 (s, 1H, –OH); FT-IR (ATR): 3420, 2883, 1703, 1584, 1438, 1395, 1256, 1175, 1082, 873, 762 cm−1.
), 5.24 (m, 1H, trans CH2), 5.16 (m, 1H, cis CH2) 3.94 (d, 2H, CH2CH–CH2–O–), 3.40 (p, 1H, –CH–OH), 3.88 (d, 2H, –O–CH2–), 3.30 (d, 2H, –CH2–N3) 3.13 (s, 1H, –OH); FT-IR (ATR): 3420, 2883, 1703, 1584, 1438, 1395, 1256, 1175, 1082, 873, 762 cm−1.
Synthesis of 1-(allyloxy)-3-azidopropan-2-yl (anthracen-9-ylmethyl) succinate (AAS, the core)
Into a 50 mL flask equipped with a magnetic stirrer, the anthracene acid compound, AAP (1.6 g, 1 eq.) was added and dissolved in 20 mL CH2Cl2. Then, AOB (2.2 g, 1.3 eq.) and DMAP (0.7 g, 1 eq.) were added to the solution. After 5 minutes of stirring, 5 mL of CH2Cl2 solution containing DCC (1.7 g, 1.3 eq.) was added to the solution and the reaction mixture was left stirring overnight. The organic phase was extracted with dilute aqueous HCl, dried over MgSO4 and filtered. The organic solvent was completely evaporated and the resulting viscous liquid was purified by column chromatography using ethyl acetate/hexane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4, v/v) as eluent. (Yield: 74%) 1H NMR (CDCl3, 250 MHz): δ = 8.49–7.48 (m, 9H, aromatic), 6.16 (s, 2H, anthracene–CH2–O), 5.84 (m, 1H, –CH), 5.27 (m, 1H, cis CH2), 5.15 (m, 1H, trans CH2), 4.02 (m, 1H, O–CH–), 3.94 (d, 2H, O–CH2–CH), 3.45 (d, 2H, –CH2–O), 3.37 (d, 2H, –CH2–N3) 2.66 (s, 4H, OC–CH2–CH2–CO); 13C NMR (CDCl3, 250 MHz): δ = 173.5 (2C, –CO), 134.2 (1C, –C) 131.1–123.8 (14C, anthracene), 117.6 (1C, CH2), 77.1 (1C, OOC–CH2), 72.1 (1C, O–CH2–), 71.6 (1C, O–CH2–vinyl), 68.0 (1C, anthracene–CH2–O), 58.4 (1C, CH2–N3), 29.0 (2C, OC–CH2–CH2–CO); FT-IR (ATR): 2943, 2791, 2101, 1728, 1592, 1447, 1088, 1021, 735 cm−1.
:4, v/v) as eluent. (Yield: 74%) 1H NMR (CDCl3, 250 MHz): δ = 8.49–7.48 (m, 9H, aromatic), 6.16 (s, 2H, anthracene–CH2–O), 5.84 (m, 1H, –CH), 5.27 (m, 1H, cis CH2), 5.15 (m, 1H, trans CH2), 4.02 (m, 1H, O–CH–), 3.94 (d, 2H, O–CH2–CH), 3.45 (d, 2H, –CH2–O), 3.37 (d, 2H, –CH2–N3) 2.66 (s, 4H, OC–CH2–CH2–CO); 13C NMR (CDCl3, 250 MHz): δ = 173.5 (2C, –CO), 134.2 (1C, –C) 131.1–123.8 (14C, anthracene), 117.6 (1C, CH2), 77.1 (1C, OOC–CH2), 72.1 (1C, O–CH2–), 71.6 (1C, O–CH2–vinyl), 68.0 (1C, anthracene–CH2–O), 58.4 (1C, CH2–N3), 29.0 (2C, OC–CH2–CH2–CO); FT-IR (ATR): 2943, 2791, 2101, 1728, 1592, 1447, 1088, 1021, 735 cm−1.
Synthesis of polystyrene with ω-xanthate functionality (PS-Xant)
Into a two necked flask equipped with a stirrer, PS-Br (0.9 g, 1 eq.), potassium xanthate (0.4 g, 5 eq.) and 8 mL DMF were added under N2 atmosphere. The reaction mixture was stirred for 12 h at room temperature and precipitated in 10 fold excess of methanol to yield PS-Xant (Mn,GPC = 2200 g mol−1, Mw/Mn = 1.20).Synthesis of polystyrene with ω-thiol functionality (PS-SH)
Into a two necked flask equipped with a stirrer, DMF (8 mL), PS-Xant (0.7 g, 1 eq.) and metallic zinc (60 mg, 3 eq.) were added under N2 atmosphere. Next, 1,2-ethandithiol (0.3 mL, 10 eq.) was added to the reaction media by a syringe. The reaction mixture was stirred for 3 h and precipitated into a slightly acidic 10 fold excess methanol/water mixture (10:1, v/v) to give a white powder solid. (Yield: 95%) Mn,NMR = 1980, Mn,GPC = 1950 g mol−1, Mw/Mn = 1.18. 1H NMR (CDCl3, 250 MHz): δ = 7.25–6.24 (m, aromatic protons of PS), 3.95 (broad, 2H, –CH2–O–CO), 3.51 (broad, 1H, –CH–S–).
Synthesis of poly(ethylene glycol) with ω-maleimide functionality (Me-PEG-MI)
Me-PEG (Mn ≈ 550 g mol−1 (0.6 g, 1 mmol)) was dissolved in 30 mL of CH2Cl2. (3) (1.0 g, 3.00 mmol), DMAP (0.1 g, 1.0 mmol) and DCC (0.5 g, 3.0 mmol) in 10 mL of dichloromethane were added to the solution in that order. The reaction mixture was stirred overnight at room temperature. The formed salt product was filtered and the remaining solution was evaporated to give a brownish viscous liquid which was purified by column chromatography over silica gel eluting first with CH2Cl2/ethylacetate (1:1, v/v), and then with methanol/CH2Cl2 (1:10, v/v). Finally, the organic phase was evaporated to give Me-PEG-MI. (Yield: 73%), Mn,NMR = 840; Mn,GPC = 1200; Mw/Mn = 1.03. 1H NMR (CDCl3, 250 MHz): δ = 6.50 (s, 2H, CHCH), 5.25 (s, 2H, CH as bridgehead protons), 4.23 (m, 4H, CH2OCO), 3.75–3.51 (m, OCH2CH2 repeating unit of PEG, CONCH2, and CH2, PEG repeating unit), 3.36 (s, 3H, PEG–OCH3), 2.87 (s, CHCH, as bridge protons), 2.61–2.56 (m, 4H, COCH2CH2CO).
Thiol–ene click reaction of PS-SH with the core (PS-N3-Ant)
PS-SH (0.8 g, 1 eq.) was dissolved in 7 mL toluene in a Pyrex tube with a stirrer under N2 atmosphere. Then core (0.9 g, 5 eq.) and bisacyl phosphineoxide (0.9 g, 10 eq.) were added to the solution in that order. The stirring solution was irradiated with a light source emitting light ranging from λ = 400–500 nm for 3 h and precipitated into hot cyclohexane to remove the excess of the core. The solvent phase was concentrated and precipitated into methanol to give polystyrene–core (PS-N3-Ant). (Yield: 88%) Mn,NMR = 2730 g mol−1, Mn,GPC = 2590 g mol−1, Mw/Mn = 1.21. 1H NMR (CDCl3, 250 MHz): δ = 8.49–7.48 (m, 9H, aromatic peaks of anthracene), 7.25–6.24 (m, aromatic protons of PS) 6.21 (s, 2H, anthracene–CH2–O), 4.03 (m, 1H, O–CH–), 3.95 (m, 2H, CH2–OCO), 3.63–3.24 (other core protons), 1.69–1.12 (m, aliphatic protons of PS); FT-IR (ATR): 3023, 2925, 2847, 2100, 1945, 1870, 1803, 1731, 1669, 1602, 1491, 1449, 1377, 1152, 1026, 752 cm−1.
CuAAC click reaction of alkyne-PCL with PS-N3-Ant (PS-PCL-Ant)
To a Schlenk tube equipped with a magnetic stirring bar, PS-N3-Ant (1 eq.), alkyne-PCL (0.8 eq.), ligand (PMDETA, 1.5 eq.), and catalyst (CuBr, 1.5 eq.) were added and dissolved in 3 mL of DMF. The tube was degassed by three freeze–pump–thaw cycles, left under vacuum, and placed in a thermostatted oil bath at 70 °C. After 24 h, the reaction mixture was diluted with THF and then passed through a column of neutral alumina to remove metal salts. The reaction mixture was concentrated and precipitated into diethyl ether/methanol mixture. The resulting polymer (PS-PCL-Ant) was further reprecipitated into diethyl ether/methanol and dried under vacuum at room temperature. (Yield: 95%) (Mn,NMR = 7310 g mol−1, Mn,GPC = 8850 g mol−1, Mw/Mn = 1.10) FT-IR (ATR): 3436, 2943, 2795, 1729, 1490, 1450, 1366, 1280, 1241, 1187, 1044, 962, 700 cm−1.DA click reaction of Me-PEG-MI with PS-PCL-Ant (PS-PCL-PEG)
Into a flask equipped with a magnetic stirrer, toluene (15 mL), PS-PCL-Ant (1 eq.) and Me-PEG-MI (1.5 eq.) were added and allowed to react for 48 h at 110 °C in the dark. After the completion of the reaction, solution was concentrated and poured into ten-fold excess methanol to give PS-PCL-PEG miktoarm star copolymer. (Yield: 97%) (Mn,NMR = 7780 g mol−1, Mn,GPC = 8720 g mol−1, Mw/Mn = 1.10).One pot synthesis of PS-PCL-PEG by the combination of CuAAC and DA
To a Schlenk tube equipped with a magnetic stirrer, PS-N3-Ant (1 eq.), alkyne-PCL (0.8 eq.), Me-PEG-MI (1.2 eq.), PMDETA (1.5 eq.), and catalyst (CuBr, 1.5 eq.) were added and dissolved in DMF (5 mL). The tube was degassed by three freeze–pump–thaw cycles, refilled with N2, and placed in a thermostatted oil bath at 110 °C. After 48 h, the reaction mixture was diluted with THF and passed through a short column of neutral alumina to remove the inorganic salts. The solvent was evaporated, and the resulting concentrated solution is poured into 10 fold excess methanol to precipitate the polymer. The polymer is further dissolved in CH2Cl2 and reprecipitated into methanol/diethyl ether mixture to gain the product (net click efficiency: 92%).Results and discussion
The core compound serving appropriate clickable sites, namely allyl, azide and anthracene functionalities, was synthesized in two sequential steps with high practical yields (Scheme 1). Thus, allyl glycidyl ether was reacted with sodium azide in ethanol/water mixture to give the allylic compound with azide and hydroxyl groups, namely 1-(allyloxy)-3-azidopropan-2-ol (AAP) nearly with quantitative yield (96%). Next, 4-(anthracen-9-ylmethoxy)-4-oxobutanoic acid (AOB), prepared in a parallel reaction, was simply linked to AAP by esterification in the presence of DCC and DMAP in dichloromethane (Yield: 74%). The structure was confirmed by 1H NMR and FT-IR analyses (see ESI, SF1 for the 1H NMR spectrum†).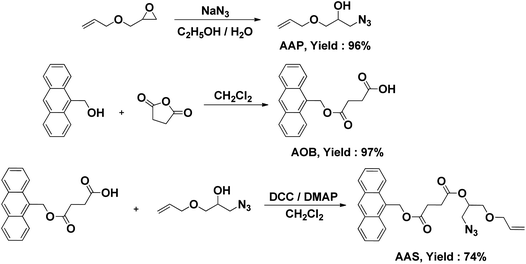 | ||
| Scheme 1 Synthesis of the core. | ||
Synthesis of polymers with well-defined clickable functionalities
ω-Thiol functionalized polystyrene (PS-SH) was synthesized in two sequential steps, starting from PS-Br which was obtained by Atom Transfer Radical Polymerization (ATRP), according to a modified procedure described previously. First, the bromide functionality was substituted with xanthate group in DMF to give PS-Xant. Then, PS-Xant was treated with 1,2-ethandithiol in the presence of metallic zinc, which suppresses the possibility of any disulfide bond formation and provides synthesis of monodisperse PS-SH. The obtained polymer is precipitated in a slightly acidic methanol/water mixture to remove any inorganic residue. The overall synthetic procedure is demonstrated in Scheme 2.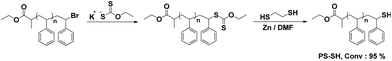 | ||
| Scheme 2 Synthesis of PS-SH. | ||
The modification of the chain end is followed by 1H NMR spectroscopy. The benzylic proton adjacent to bromide functionality (4.41 ppm) is observed to completely disappear, whereas a new peak at 3.51 ppm corresponding to the benzylic proton near the thiol functionality emerges. The efficiency of functionalization conversion as determined by comparing the integrated signals of these protons to the total aromatic protons of polystyrene is found to be 95% (Fig. 1a).
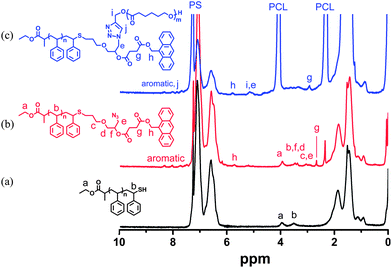 | ||
| Fig. 1 1H NMR spectra of (a) PS-SH, (b) PS-N3-Ant and (c) PS-PCL-Ant. | ||
For the CuAAC click reaction, alkyne functional poly(ε-caprolactone) (alkyne-PCL) was prepared using the Ring Opening Polymerization (ROP) technique. Polymerization using propargyl alcohol as initiator and tin(II) 2-ethylhexanoate as catalyst afforded alkyne-PCL with narrow molecular weight distribution (Table 1). It should be noted that in the process, the possibility of a transesterification reaction yielding a polymer with uncontrolled molecular weight characteristics is prevented, since the polymerization media is diluted by using toluene as solvent.
| Polymer | Yield (%) | M n,NMR a/g mol−1 | M n,GPC b/g mol−1 | M w/Mnb |
|---|---|---|---|---|
| a Determined by 1H NMR analysis. b Determined by gel permeation chromatography (GPC) using polystyrene standards. c Conversion of monomers as determined gravimetrically. d Yields of the respective reactions determined by 1H NMR analysis. | ||||
| PS-SH | 95d | 1910 | 2150 | 1.18 |
| Alkyne-PCL | 12c | 5050 | 4700 | 1.17 |
| Me-PEG-MI | 70d | 740 | 590 | 1.10 |
As the core introduces anthracene as the third clickable functionality, a polymer bearing a dienophile chain-end is designed and synthesized. For this purpose, a compound carrying both maleimide and carboxylic acid functional groups was synthesized and attached to Me-PEG-OH by a simple esterification reaction to yield Me-PEG-MI (Scheme 3).
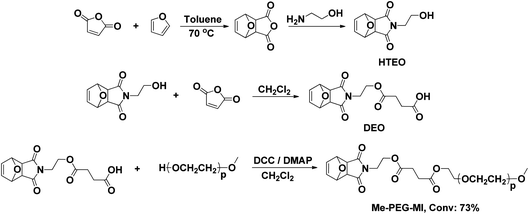 | ||
| Scheme 3 Synthesis of Me-PEG-MI. | ||
The yields of polymerization and functionalization reactions as well as the molecular weight characteristics of the resulting polymers are presented in Table 1.
Grafting of polymers onto the core: click reactions
Since thiol–ene reactions are usually limited to occur between molecules of low molecular weights, the first step of grafting onto processes should be the thiol–ene click reaction. Thus, PS-SH is clicked to the allyl group of the core using bis-(2,4,6-trimethylbenzoyl)-phenylphosphine oxide (BAPO) as the photo-labile radical source (Scheme 4). BAPO is deliberately selected, due to its strong UV-absorbance at above 400 nm where the anthracene chromophore of the core is completely transparent (ESI, SF2 and SF3†).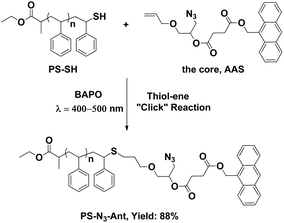 | ||
| Scheme 4 Grafting of PS-SH onto the core by thiol–ene “click” reaction. | ||
The 1H NMR spectrum of the resulting polymer proves the success of the thiol–ene click reaction (Fig. 1b). Clearly, the peak associated with the benzylic proton adjacent to the thiol functionality completely disappeared and new peaks corresponding to the core compound rises. The yield of the reaction is calculated by comparing the integrated area of the anthracene protons with the total area of polystyrene and found to be realized with 88% conversion.
The attachment of the core is further confirmed by FT-IR analysis. The polymer is found to display characteristic azide and carbonyl stretching bands around 2100 and 1730 cm−1, which verify the success of the click process (vide infra).
Next, alkyne-PCL is grafted onto the core by CuAAC click reaction using CuBr/PMDETA as the catalyst system to give PS-PCL-Ant as shown in Scheme 5.
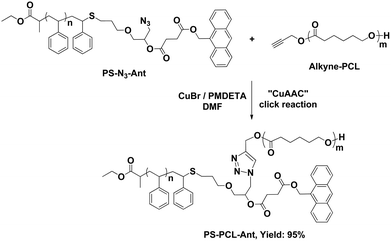 | ||
| Scheme 5 CuAAC click of alkyne-PCL with PS-N3-Ant. | ||
The reaction is monitored by 1H NMR spectroscopy. Fig. 1c shows the characteristic protons of PCL segment without any signals of CH2 protons of the propargyl group at 4.69 ppm. The appearance of the peak associated with the triazole proton (proton j) at 7.92 ppm further confirms the success of the click reaction. By comparing the ratio of the integrated areas of i and e protons with the total aromatic peaks of PS (and/or with the characteristic broad peak of PCL around 2.3 ppm), the efficiency of CuAAC is calculated to occur with 95% yield.
The CuAAC click reaction is also monitored by FT-IR measurements (Fig. 2). The FT-IR spectrum of the obtained polymer demonstrates the presence of both polymeric segments. Peaks at 1729, 1366, 1280, 1187, 1044, and 962 cm−1 are arising from the PCL segment, whereas transitions at 1490, 1450, 700 cm−1 are due to the PS component. After click reaction, the peak at 2100 cm−1 associated with the –N3 stretching is not observed, confirming the quantitative attachment of alkyne-PCL by the click reaction.
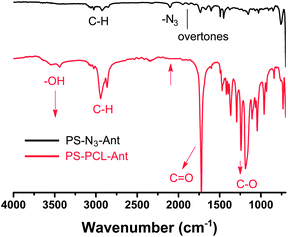 | ||
| Fig. 2 FT-IR spectra of PS-N3-Ant and PS-PCL-Ant. | ||
The last grafting onto process is carried out by reacting PS-PCL-Ant with Me-PEG-MI using DA click reaction (Scheme 6).
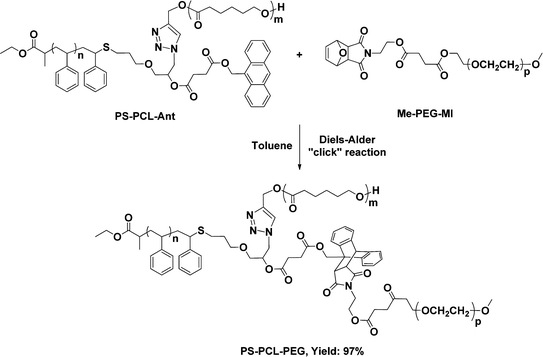 | ||
| Scheme 6 DA click of PS-PCL-Ant with Me-PEG-MI. | ||
Fig. 3 demonstrates the 1H NMR spectrum of the final PS-PCL-PEG miktoarm star copolymer. In the figure where the integrated areas are also included, the characteristic peaks of the three segments are detected. Moreover, the appearance of characteristic PEG protons at 3.64 ppm and the absence of the aromatic protons belonging to the anthracene ring evidence nearly quantitative final DA click reaction (ca. 97%). The yield of the reaction is calculated by comparing the integrated area of total PEG protons with the aromatic protons of the PS segment.
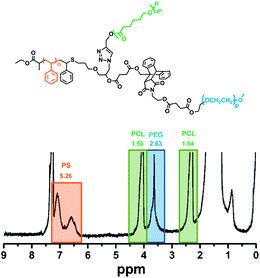 | ||
| Fig. 3 1H NMR spectrum of the PS-PCL-PEG star copolymer. | ||
Fig. 4 shows GPC traces of the polymers at different stages. Clearly, all polymers exhibit unimodal curves with narrow molecular weight distribution characteristics, evidencing that chain scissions or side reactions can be excluded through the whole process. After clicking of the PCL segment, the GPC trace shifts to higher molecular weights, whereas incorporation of the PEG segment reflects a slightly lower molecular weight determination at the final stage. This can be attributed to the strong dipolar interactions between the hydrophilic PEG segment and the stationary phase of the GPC column.
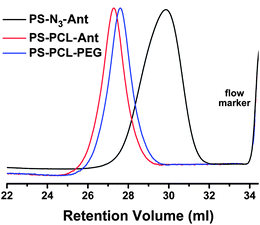 | ||
| Fig. 4 GPC traces of PS-N3-Ant, PS-PCL-Ant and PS-PCL-PEG. | ||
The yields of the respective click reactions and the molecular weight of the polymers at various stages are presented in Table 2.
In order to demonstrate the possibility of carrying out CuAAC and DA reaction simultaneously, a one-pot double click reaction is performed. Thus, PS-N3-Ant, alkyne-PCL and Me-PEG-MI are reacted together in the same reaction media, at temperatures where DA reaction is enabled (110 °C). At 48 h of reaction time, both the arms are observed to efficiently click to each other, verifying the modularity of these reactions. (The net click efficiency is calculated by multiplying the efficiencies of respective click reactions and found to be 92% using the 1H NMR data.)
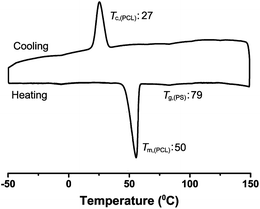
The thermal properties of the star copolymer are analyzed by means of DSC measurements (Fig. 5). The sharp endotherm and the strong exotherm around 50 °C and 27 °C correspond to the melting (Tm) and the crystallization temperatures (Tc) of the PCL segment, respectively. The detected glass transition temperature (Tg) around 79 °C belongs to the PS arm of the star copolymer. This value is lower than the expected value (95–105 °C) probably due to short PS chain and/or the miscibility of the PS and PEG segments in this molecular weight composition.
Conclusion
In conclusion, a new compound possessing different clickable functional groups, namely alkene, azide and anthracene, was synthesized in a two-step procedure with high yields. These groups were clicked with polymers with convenient clickable functionalities. As a result, an ABC type heteroarm star copolymer is synthesized by combining the three click reactions: thiol–ene, CuAAC and DA. The obtained organic structures as well as polymeric blocks were evidenced by spectral analyses. Provided that the related functional polymers are obtainable, the approach described here may open new pathways for the preparation of complex macromolecular structures possessing synthetic polymers and biomolecules.Acknowledgements
The authors thank Istanbul Technical University for financial support. One of the authors (YY) thanks Turkish Academy of Sciences (TUBA) for partial financial support.References
- N. Hadjichristidis, M. Pitsikalis, S. Pispas and H. Iatrou, Chem. Rev., 2001, 101, 3747–3792 CrossRef CAS.
- N. Hadjichristidis, H. Iatrou, M. Pitsikalis and J. Mays, Prog. Polym. Sci., 2006, 31, 1068–1132 CrossRef CAS.
- J. S. Wang and K. Matyjaszewski, Macromolecules, 1995, 28, 7901–7910 CrossRef CAS.
- T. E. Patten and K. Matyjaszewski, Adv. Mater., 1998, 10, 901–915 CrossRef CAS.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS.
- D. Benoit, V. Chaplinski, R. Braslau and C. J. Hawker, J. Am. Chem. Soc., 1999, 121, 3904–3920 CrossRef CAS.
- T. Fukuda, T. Terauchi, A. Goto, K. Ohno, Y. Tsujii, T. Miyamoto, S. Kobatake and B. Yamada, Macromolecules, 1996, 29, 6393–6398 CrossRef CAS.
- D. Greszta and K. Matyjaszewski, Macromolecules, 1996, 29, 7661–7670 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- B. Gacal, H. Durmaz, M. A. Tasdelen, G. Hizal, U. Tunca, Y. Yagci and A. L. Demire, Macromolecules, 2006, 39, 5330–5336 CrossRef CAS.
- J. F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018–1025 CrossRef CAS.
- D. Fournier, R. Hoogenboom and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1369–1380 RSC.
- B. Helms, J. L. Mynar, C. J. Hawker and J. M. J. Frechet, J. Am. Chem. Soc., 2004, 126, 15020–15021 CrossRef CAS.
- J. F. Lutz, H. G. Borner and K. Weichenhan, Macromol. Rapid Commun., 2005, 26, 514–518 CrossRef CAS.
- O. Altintas, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5699–5707 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- B. M. Trost, Science, 1991, 254, 1471–1477 CAS.
- M. A. Tasdelen, Polym. Chem., 2011, 2, 2133–2145 RSC.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS.
- B. S. Sumerlin and A. P. Vogt, Macromolecules, 2010, 43, 1–13 CrossRef CAS.
- K. L. Killops, L. M. Campos and C. J. Hawker, J. Am. Chem. Soc., 2008, 130, 5062–5064 CrossRef CAS.
- L. M. Campos, K. L. Killops, R. Sakai, J. M. J. Paulusse, D. Damiron, E. Drockenmuller, B. W. Messmore and C. J. Hawker, Macromolecules, 2008, 41, 7063–7070 CrossRef CAS.
- M. J. Kade, D. J. Burke and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 743–750 CrossRef CAS.
- N. ten Brummelhuis, C. Diehl and H. Schlaad, Macromolecules, 2008, 41, 9946–9947 CrossRef CAS.
- S. P. S. Koo, M. M. Stamenović, R. A. Prasath, A. J. Inglis, F. E. Du Prez, C. Barner-Kowollik, W. Van Camp and T. Junkers, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1699–1713 CrossRef CAS.
- A. Dondoni, Angew. Chem., Int. Ed., 2008, 47, 8995–8997 CrossRef CAS.
- L. Nurmi, J. Lindqvist, R. Randev, J. Syrett and D. M. Haddleton, Chem. Commun., 2009, 2727–2729 RSC.
- C. Boyer and T. P. Davis, Chem. Commun., 2009, 6029–6031 RSC.
- J. Sun and H. Schlaad, Macromolecules, 2010, 43, 4445–4448 CrossRef CAS.
- B. Gacal, H. Akat, D. K. Balta, N. Arsu and Y. Yagci, Macromolecules, 2008, 41, 2401–2405 CrossRef CAS.
- A. Dag, H. Durmaz, E. Demir, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6969–6977 CrossRef CAS.
- G. Temel, B. Aydogan, N. Arsu and Y. Yagci, Macromolecules, 2009, 42, 6098–6106 CrossRef CAS.
- B. Iskin, G. Yilmaz and Yagci, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2417–2422 CrossRef CAS.
- A. S. Goldmann, A. Walther, L. Nebhani, R. Joso, D. Ernst, K. Loos, C. Barner-Kowollik, L. Barner and A. H. E. Mueller, Macromolecules, 2009, 42, 3707–3714 CrossRef CAS.
- X. G. Lei and J. A. Porco, Org. Lett., 2004, 6, 795–798 CrossRef CAS.
- G. Mantovani, F. Lecolley, L. Tao, D. M. Haddleton, J. Clerx, J. Cornelissen and K. Velonia, J. Am. Chem. Soc., 2005, 127, 2966–2973 CrossRef CAS.
- J. S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- R. Hoogenboom, B. C. Moore and U. S. Schubert, Chem. Commun., 2006, 4010–4012 RSC.
- C. Jerome and P. Lecomte, Adv. Drug Delivery Rev., 2008, 60, 1056–1076 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1py00352f |
| This journal is © The Royal Society of Chemistry 2011 |