Main-chain photochromic conducting polymers
Christopher P.
Harvey
and
John D.
Tovar
*
Department of Chemistry and Department of Materials Science and Engineering, Johns Hopkins University, 3400 N. Charles St. (NCB 316), Baltimore, MD 21218, USA. E-mail: tovar@jhu.edu
First published on 26th August 2011
Abstract
The optoelectronic properties of photochromic materials change in response to exposure to certain wavelengths of light. These alterations arise from specific electronic reorganization of conjugation pathways due to photochemically-triggered cyclizations and conformational changes. Polymers with photochromic switches pendent to the backbone retain monomer-like optoelectronic photoswitching responses while incorporation of photochromic switches directly into a conjugated backbone allows for greater, and often more dramatic, influence on the polymer optoelectronic properties. This ability for light-controlled variation of optoelectronic properties has driven research in the field of main-chain photochromic conducting polymers, and recent developments in this area will be discussed.
 Christopher P. Harvey | Christopher Harvey received his Bachelor of Science degree in Chemistry from the Pennsylvania State University in 2004. He then began graduate studies at Johns Hopkins University where he joined the research group of Prof. J. D. Tovar in 2005. In the Tovar group, he began studying segmented polyurethane elastomers as mechanochromic materials and later branched into the investigation of photochromic switching systems. His primary research interest is stimuli-responsive materials. |
 John D. Tovar | J. D. Tovar, a native of Iowa, completed undergraduate studies in chemistry at the University of California, Los Angeles followed by doctoral studies in organic chemistry the Massachusetts Institute of Technology and postdoctoral training at Northwestern University. He has been at Johns Hopkins since 2005 where his research examines synthetically complex organic semiconductors spanning small molecule, polymeric and bioelectronic supramolecular structures. (Photo credit: Will Kirk, JHU.) |
Introduction
Photochromic molecules undergo reversible color changes in response to irradiation with visible and/or ultraviolet light, and they are found in a variety of natural pigment systems and non-natural electronic applications.1 These color changes are the result of cyclizations or ring opening rearrangements within the molecules which alter their optoelectronic properties by modifying the available conjugation pathways. Photochromic compounds may revert to their original forms either thermally or in response to irradiation with a different wavelength of light. One natural photochromic protein, bacteriorhodopsin, is involved in photosynthesis in halobacteria.2Photoisomerization of a retinylidene residue within this protein is coupled with proton conduction causing a gradient used by the bacteria to form ATP. Spiropyrans are another class of photochromic switches and they have also been incorporated into polymers creating stress-responsive materials that exhibit mechanochromic responses to mechanical deformation.3,4Photochromic molecules comprise one subset of the broad family of stimuli-responsive materials whereby multiple external stimuli can lead to defined alterations of materials properties, most commonly for photochromic materials in the context of chromic or electronic changes. Photochromic switches may be useful for optoelectronic devices and in particular for optical memories and data storage due to the fatigue-resistance often engineered into photochromic molecules; that is, the resistance to thermally-induced reversions among ring-opened and ring-closed forms.5 This allows for the application of a specific optical stimulus to “write” information that can be read non-destructively and only “erased” with another specific optical stimulus. Photochromic systems have also been employed more recently in the context of light-controlled catalysis,6 Lewis basicity,7 and liquid crystallinity,8 as well as to realize molecular electronic devices with light-tunable conductance,9 and even to regulate biological functions.10,11 One area that has been less established is the prospect for using light as a stimulus to directly impact electrical transport properties with an orthogonal energy currency (photochemical vs. electrical). This review will briefly introduce the general topic of photochromicity in the context of the established dithienylethene photochromic switching motif and then discuss the current photochromic conducting polymer state-of-the-art and prospects for future advancement.
Examples of dithienylethene (DTE) photochromic switches
Dithienylethenes are one class of photochromic compounds that undergoes an intramolecular cyclization in response to specific wavelengths of UV light. This 6-pi electrocyclization takes place between the pendent thiophene groups. The dithienylethene switching motif is useful because it exhibits chemical and thermal stability and resists photoswitching fatigue.5 As shown in Fig. 1, the pendent thiophene groups in dithienylethenes may exist as either anti-parallel or parallel rotamers.12,13 The conrotatory photocyclization which leads to photochromic switching can only proceed from the anti-parallel conformation.13 Because only one of the two rotamers is photochemically active the quantum yield for the overall system is lowered by the presence of the inactive isomer. The open ring form of the photochromic switch (1o) does not absorb visible light (λmax 234 nm) due to the limited conjugation pathway.14 Although the thiophene appendages of 1o are mutually conjugated to a double bond, these moieties are not coplanar, and linkage at the 3-position does not foster extended conjugation. In the closed ring form (1c), the extended conjugation within the planarized oligoene pathway causes the system to absorb in the visible region (λmax 534 nm).14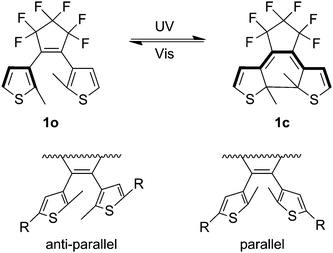 | ||
| Fig. 1 Top: change in conjugation pathway due to photochromic switching. Modified with permission from ref. 14 Copyright 1999 Elsevier Science S.A. Bottom: rotamers of dithienylethenes. Modified with permission from ref. 12 Copyright 2007 The Royal Society of Chemistry. | ||
Materials with photochromic switches pendent to the main polymer backbone can be synthesized by combining photochromic switching motifs with moieties which allow for facile polymerization. In these materials, the photochromic switching occurs in groups removed from the polymer backbone which leads to polymeric materials that respond as the isolated molecular photochromic switch. Branda reported compound 2 containing both a dithienylethene photochromic switch and a strained tricyclic group which allowed for ring-opening metathesis polymerization (ROMP) to form polymer 3 (Fig. 2).15 This material exhibited photochromic properties in solution with initially colorless ring-open 3 (λmax 248 nm) undergoing cyclization to the pink colored ring-closed form (λmax 512 nm) under UV light irradiation. The solution returned to the colorless state after irradiation with visible light (λ > 434 nm). Films of polymer 3 also exhibited this coloration/de-coloration behavior indicating their function as a solid-state polymer switch that presented the spectral properties of the molecular chromophore.
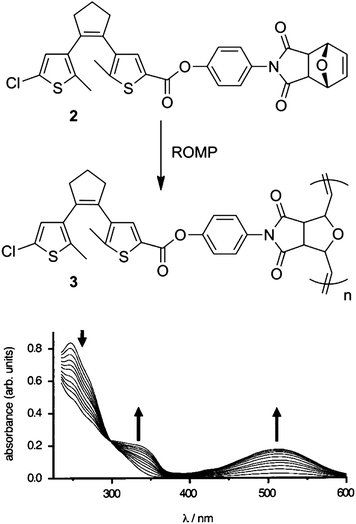 | ||
| Fig. 2 Top: ROMP polymerization of monomer 2 to form polymer 3. Bottom: UV-vis response of polymer 3 to irradiation with 254 nm light for 2–40 s in THF solution (arrows indicate the trends in absorption with increasing irradiation). Modified with permission from ref. 15 Copyright 2000 American Chemical Society. | ||
The change in conjugation pathway which accompanies photochromic switching modifies the optical and electronic properties of the material due to the extended conjugation through the closed form relative to the open form. Lehn reported the first hexafluorocyclopentene-based dithienylethene switch substituted with positively charged pyridinium rings, compound 4.16 As shown in Fig. 3, the open-form 4o exhibited no electrochemical response during cyclic voltammetry (CV). However, CV of closed-form 4c showed a reversible one electron reduction. These CV results indicate that the electronic properties of the system are controlled by the state of the photochromic switch.
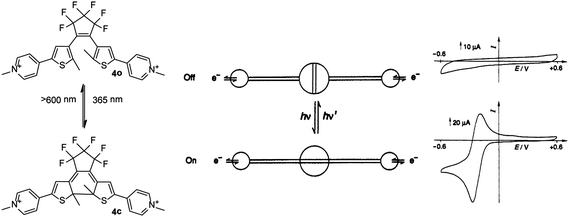 | ||
| Fig. 3 Left: pyridinium substituted dithienylethene photochromic switch 4 in the open (4o) and closed (4c) forms. Center: representation of electronic on/off switching between the open and closed forms. Right: CV of 4o (top) and 4c (bottom). Modified with permission from ref. 16 Copyright 1993 The Royal Society of Chemistry. | ||
The change in optoelectronic characteristics of photochromic switching compounds can allow control of properties such as the ability for electrochemical polymerization. Feringa reported compound 5, a terthiophene appended hexafluorocyclopentene-based photochromic switch, which would undergo electrochemical polymerization in the open form (5o) but not in the closed form (5c) as shown in Fig. 4.17 During the electrochemical experiments, the terthiophene appendages of 5o form radical cations which undergo homocoupling to produce polymer 6. The electrochemical response of 5c is more confined to the central polycyclic aromatic and the radical cation intermediate does not delocalize as in 5o. Since the stable radical cation does not extend to the termini, this system does not undergo electrochemical polymerization. Cyclic voltammetry shows the polymer growth for 5o and a reversible oxidation without polymerization for 5c.
 | ||
| Fig. 4 Top: terthiophene based diarylethene 5 and electropolymerization of 5o to form 6. Bottom Left: multiple CV traces of 5o showing the successful electropolymerization. Bottom Right: CV of 5c. Modified with permission from ref. 17 Copyright 2008 American Chemical Society. | ||
DTE-based main-chain polymers
Backbone photochromic polymers where the switching unit is also part of the main conjugation pathway were reported by Zerbi in 1999.14,18 By having the photochromic switches directly in the conjugated backbone of the polymer, the change in conjugation pathway associated with switching can have a greater impact on the overall optoelectronic properties of the system. As shown in Fig. 5, this material was a polymerized version of 1o. Like 1o, UV irradiation of 7o led to ring closure forming 7c. The absorption λmax of 7o occurs at approximately 320 nm and decreases after UV irradiation while a new peak grows in at 620 nm corresponding to 7c.14,18 Even when a sample of polymer 7 is irradiated to the photostationary state forming the maximum possible amount of 7c there is still a substantial absorption from 7o.18 The continued presence of the 7o absorption indicates that not all of the switching units are closed in the photostationary state. Monomer 1 and polymer 7 have different quantum yields for photochromic switching in solution. The quantum yield for closure of 1o is 21% whereas it is 86% for the 7o.14 Monomer 1c has a quantum yield for opening of 13% versus 0.15% for polymer 7c.14 This shows that ring closure within the polymer is more facile than ring opening. CV experiments conducted with these materials show different results for 7oversus7c. Polymer 7o has no reversible electrochemical response and decomposes during the experiment.14,18 However, 7c does not decompose and has a reversible oxidation peak at approximately 0.65 V.14,18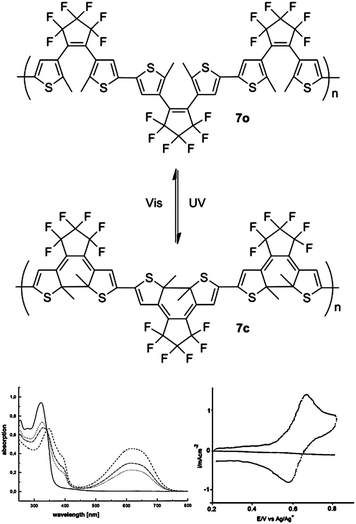 | ||
| Fig. 5 Top: photochromic switching of polymer 7. Bottom Left: UV-vis absorption spectra of 7o in CH2Cl2 solution (solid line), after irradiation with 313 nm light for 20 s (dotted line) and 60 s (dotted-dashed line), and in the photostationary state (dashed line) showing increased low-energy absorption attributed to 7c. Bottom Right: CV of 7c. Modified with permission from ref. 18 Copyright 1999 John Wiley & Sons, Inc. | ||
Also in 1999, Irie reported photochromic switches with benzothiophene-containing molecules.19 Incorporation of this switch into polymers was accomplished by palladium catalyzed Suzuki cross coupling of dioctylfluorene derivatives with a functionalized version of the photochromic switching core 8.19,20 Films of the resulting polymer 9 were pale yellow and became dark red after UV irradiation. In solution, UV irradiation causes a decrease in the intensity of the UV-vis absorption peak from the open-ring form of 9 at 340 nm and the appearance of two new lower energy absorptions corresponding to closed-ring 9 at 400 nm and 560 nm (Fig. 6). When a polymer film was irradiated, the initial absorption peak did not decrease as much as when the polymer was in solution. This indicates that the photochromic switches close less readily within the polymer film. There is also an effect on the photoluminescence spectrum of 9 upon photoisomerization showing a decrease in photoluminescence intensity of approximately one-third upon photoisomerization forming closed-ring 9. When open-form 9 is spin coated into a film, photoisomerization to the closed form by UV irradiation to the photostationary state prior to the photoluminescence measurement caused near-complete quenching of photoluminescence. The electrical conductivity of 9 is also affected by the state of the photochromic switch. Open-ring 9 has a conductivity of 5.3 × 10−13 S cm−1 whereas the irradiated closed-ring form has a conductivity of 1.2 × 10−12 S cm−1. The higher conductivity of the closed-ring form is attributed to the extended conjugation pathway throughout the polymer backbone. The conductivity of the open-ring form of 9 can be increased by doping with iodine vapor. The doped polymer has a conductivity of 1.4 × 10−8 S cm−1 which is five orders of magnitude higher than the undoped material. The effect of iodine doping on the closed form of 9 was not reported.19
 | ||
| Fig. 6 Benzothiophene substituted diarylethene photochromic switch monomer 8 (top) and the related polymer 9 (middle). UV-vis absorption spectra (bottom) of 9 in THF solution (left) and as a spin coated film (right) in the original state (solid lines) and at the photostationary state (dashed lines) under irradiation with 313 nm light. Modified with permission from ref. 19 Copyright 1999 The Chemical Society of Japan. | ||
Inspired by these seminal results, there has been a tremendous interest in the synthesis of main-chain photochromic polymers. Chart 1 illustrates some of the reported materials based on the dithienylethene photoswitch. During the final preparation of this review, Tian and coworkers published a related review also focusing on photochromic polymers.21 This present contribution will provide more discussion of the physical properties of the main-chain photochromic materials and serves as a complement to Tian's prior independent review.
 | ||
| Chart 1 Dithienylethene photochromic switch based polymers (shown in ring-open form).22–25 | ||
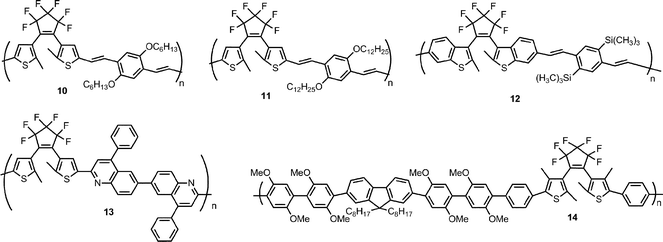
In 2004, Zerbi reported polymers 10 and 11 which included both hexafluorocyclopentene-based dithienylethene switching units and dialkoxyphenylene derivatives.22 These polymers were synthesized by a Horner polymerization of dialkoxy diphosphonate xylylene and dithienylethene dialdehyde to create soluble phenylene vinylene polymers. As shown in Fig. 7, the UV-vis absorption profiles of the materials were similar with either alkoxy group. The open form of either polymer had an absorption maximum around 420 nm. After UV irradiation, both materials showed a new absorption near 680 nm resulting from the closed form of the polymer. A substantial absorption from the open form was still present after irradiation indicating incomplete photoisomerization. In solution, CV of polymer 10 in the closed-ring form showed a reversible oxidation peak near 0.8 V but the open ring isomer did not have a reversible CV response.
 | ||
| Fig. 7 UV-vis absorption spectrum of (left) open-ring 10 (solid line) and a mixture of open- and closed-ring 10 after UV irradiation (dashed line) in CHCl3 solution and (right) solid state 11 at different irradiation times. Modified with permission from ref. 22 Copyright 2004 John Wiley & Sons, Inc. | ||
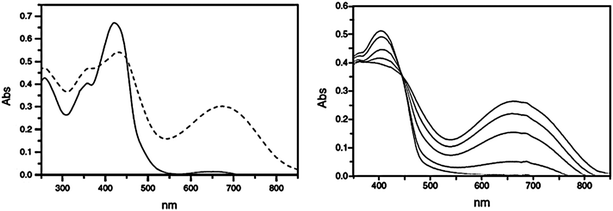
Kim reported polymer 12 containing benzothiophene appended hexafluorocyclopentene-based photochromic switches and trimethylsilyl substituted phenylene vinylene units.23 The thin-film conductivity of pristine 12 can be switched in response to UV or visible light irradiation, as shown in Fig. 8. The conductivity of open-form 12 is 3 × 10−9 S cm−1 while that in the closed form is 2.5 × 10−8 S cm−1. These pristine conductivities are higher than those reported for other diarylethene based polymers, but related measurements of doped polymers were not reported. The current–voltage graphs show the increased current flow in the closed-ring form versus the open-ring form. The current–voltage response could be cycled at least 15 times with alternating UV and visible light without showing substantial degradation. The difference in conductivity between the open-ring form and the closed-ring form allows for the possibility of using lithography to print electrodes with electrical properties that can be controlled by UV or visible light exposure. A photograph of one such printed electrode shows bright areas corresponding to regions of the open-ring isomer and dark regions containing the closed-ring isomer formed after irradiation with UV light.
 | ||
| Fig. 8 Left: I–V plots of a cell containing a film of 12 after UV and visible light exposure. (i) as prepared under UV irradiation; (ii) after irradiation with visible light for 30 min; (iii) 2nd cycle, after UV irradiation for 30 min; and (iv) 2nd cycle, after irradiation with visible light for 30 min. Right: photograph of an electrode image written on a colored film of 12 through irradiation with a 532 nm laser and the use of a mask. The dark area represents the masked closed-ring region, and the bright areas the unmasked open-ring region. The gap between the nearest two bright bars is 5 μm. Modified with permission from ref. 23 Copyright 2006 The Royal Society of Chemistry. | ||
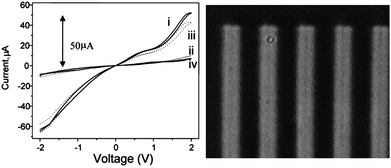
In 2005, Ko reported materials prepared by Friedländer condensation of a diacetyl substituted hexafluorocyclopentene-based dithienylethene switching unit and 3,3′-dibenzoylbenzidine which ultimately paired the switching unit with phenylquinoline units.24 Polyquinolines were chosen for their thermal stability and their ability to form thin films. Polymer 13 has two low wavelength absorptions in the open form near 290 nm and 390 nm. After irradiation, there is a broad absorption at longer wavelength centered about 650 nm. In solution the ring closure reaction occurs with quantum yields over 60% while the corresponding ring opening occurs with quantum yields under 1% as found with other polymers containing dithienylethene-based photochromic switches. As shown in Fig. 9, the current–voltage response of the material changes for the closed ring form versus the open ring form showing that they have different electronic properties. A film of neat polymer 13 shows an increase in current upon irradiation with UV light to form the closed-ring isomer. This behavior is also observed in polystyrene blends and is attributed to the extended conjugation pathway present in the closed-ring form. The conductivity of the neat polymer film is about an order of magnitude higher than that of the polystyrene blend.
 | ||
| Fig. 9 Applied voltage dependence of the current for a photocell containing (left) polystyrene (90 wt %) and 13 (10 wt %) and (right) the pure polymer film of 13 upon irradiation with 365 nm light (colored, dotted line) and 532 nm (bleached, solid line). Modified with permission from ref. 24 Copyright 2005 American Chemical Society. | ||
Polymers featuring dithienylethene switches in the main-chain typically exhibit slow photochromic response with limited switching conversion. The poor response has been attributed to intramolecular interactions between the dithienylethene units which inhibited the photochromic switching action of the interacting units due to the large changes in macromolecular conformations required. In 2005, Irie reported polymer 14 composed of hexafluorocyclopentene-based switches and oligophenylene-fluorene spacers.25 Adding spacer units to the polymer may suppress any detrimental intramolecular interactions between the switching units allowing for better photochromic performance.
A film of polymer 14 was spin cast from an irradiated solution with approximately 70% of the switching units in the closed form.25 Casting the film with the switches closed ensures that the switch units are not trapped in an inactive form within the polymer film. When this film was subsequently irradiated with visible light, the initial color was bleached. Upon subsequent irradiation with UV light, the polymer re-cyclized to an extent somewhat less than the originally cast state. UV analysis of the irradiated film showed that the absorption from the closed form reaches 69% of its original level. These results indicate that about 48% of the switches in the polymer film are susceptible to solid state photoswitching. As shown in Fig. 10, the open and closed forms of 14 have different conductivities. The closed-ring form of the polymer has a higher conductivity (approximately 1 × 10−15 S cm−1) than the open-ring form (>1 × 10−16 S cm−1) near room temperature without doping. This allows the conductivity of the system to be cycled with alternating exposure to UV and visible light, although the magnitude of the conductivity difference decreased during subsequent cycles.
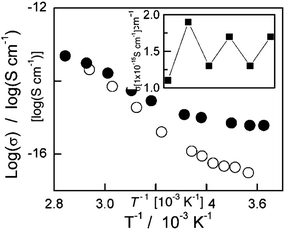 | ||
| Fig. 10 Temperature dependence of the electrical conductivity of a film of polymer 14. Solid circles: initial colored state from UV irradiated solution. Empty circles: after photobleaching with visible-light irradiation. Inset: reversible change in electrical conductivity, with alternate irradiation by UV and visible light at 280 K. Reprinted with permission from ref. 25 Copyright 2005 John Wiley & Sons, Inc. | ||
Other examples of main-chain photochromic materials
In principle, any photochromic switching unit conjugated within a pi-electron polymer should lead to comparably alterable conductivity and optoelectronic properties. In 2000, Marsella reported a conducting polymer containing the dimethyldihydropyrene photochromic switch previously investigated by Mitchell.26–28 This switching motif interconverts between a dimethyldihydropyrene ring-closed isomer and an open-ring cyclophanediene form.27,28 When the switch is irradiated with visible light above a certain wavelength (λ > 400 nm for 15c), ring opening occurs, and subsequent irradiation with UV light causes reversion to the ring-closed form.26 The dimethyldihydropyrene core was incorporated into polymers by Suzuki cross coupling polymerization of a dihalogenated switch core with bithiophene bisboronic acid. The closed form has a conjugation pathway through the switching core while the open form has a more localized electronic structure. A thin film of 15c was conductive in the potential window between 0.4 V to slightly over 1.0 V as measured on interdigitated electrodes (Fig. 11).26,29 Solid state switching could not be observed due to the slow switching speed of the system resulting from the low quantum yield, which was determined (Φ = 0.02) for the photoswitch monomer.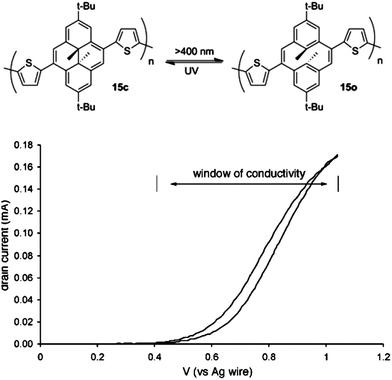 | ||
| Fig. 11 Top: photochromic switching of dimethyldihydropyrene-based polymer 15. Bottom: relative conductivity (reported as drain current) of a thin film of 15c as a function of gate voltage. Reprinted with permission from ref. 26 Copyright 2000 American Chemical Society. | ||
Spirobenzopyrans are another class of photochromic materials. The ring closed spirobenzopyran form is colorless while the ring open merocyanine form is colored. Ng used Sonogashira palladium catalyzed cross coupling reactions to incorporate this type of photochromic switch into the backbone of poly(p-phenylene ethynylene) (PPE) polymer 16.30 As shown in Fig. 12, the change in the UV-vis spectrum of 16 after irradiation is smaller than that observed for other systems such as the dithienylethene based materials. This limited response may be the result of relatively few of the photoswitches undergoing ring-opening within the polymer even in solution.
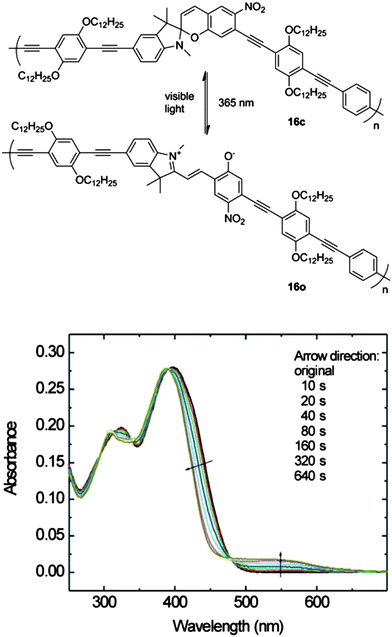 | ||
| Fig. 12 Top: photochromic switching motif of spirobenzopyran functionalized polymer 16. Bottom: UV-vis spectral response of 16 to UV irradiation over 640 s in CHCl3 solution. Reprinted with permission from ref. 30 Copyright 2006 Georg Thieme Verlag. | ||
Future directions
Incorporation of photochromic switches into materials offers the ability to control optoelectronic properties with light irradiation. The extent to which these properties can be varied is determined by the different switching motifs used but also by the other components in the matrix. Diverse properties are observed for systems containing similar dithienylethene-based photochromic switches due to the impact of the linking units incorporated into these polymers. Despite the advancements in solid-state conjugated photochromic polymers, these materials in general still suffer from fairly slow if not irreversible responses as well as overall low electrical conductivities. The ability to tune the electrical properties regardless of relative magnitude may be useful in many applications, but others may require a greater extent of discrimination (such as greater “on-off” ratios). Future work with photochromic polymers will need to be focused on increasing the extent of switching response, and the speed with which this response is achieved.One area where materials design may be able to play a role is the development of new switching motifs that may minimize macromolecular re-organization in the solid-state. Although many solution-based photochromic materials can achieve high cyclization quantum yields, mirroring this behavior in the solid-state requires dramatic conformational changes that may simply be unobtainable. Materials systems whereby the thin-film solid-state polymer matrix imparts minimal resistance to the conformational motion necessary to achieve light-triggered electrocyclic chemistry will provide an important solution to this problem. This may be realized through the development of new switching core motifs that are conjugated into polymer backbones in new ways. In this regard, our group is studying new polymer connectivities whereby photochromic switching occurs in a manner orthogonal to the conjugated polymer backbone. As foundation for this work, we recently reported the impacts of “evolved aromaticity” orthogonal to a conjugated polymer main-chain and how this can influence the electronics of the neutral and conductive forms of the polymers.31 We are designing new photochromic switching motifs that might be able to locally “make or break” such evolved aromaticity without the requirements for main-chain macromolecular conformational changes, and these alternative photochromic switching designs will be disclosed in the near future.
Acknowledgements
Our ongoing research on photochromic polymers is generously supported by Johns Hopkins University and the National Science Foundation (CAREER, DMR-0644727).References
- H. Bouas-Laurent and H. Dürr, Pure Appl. Chem., 2001, 73, 639–665, DOI:10.1351/pac200173040639
.
- N. Hampp, Chem. Rev., 2000, 100, 1755–1776, DOI:10.1021/cr980072x
- S. L. Potisek, D. A. Davis, N. R. Sottos, S. R. White and J. S. Moore, J. Am. Chem. Soc., 2007, 129, 13808–13809, DOI:10.1021/ja076189x
- D. A. Davis, A. Hamilton, J. Yang, L. D. Cremar, D. V. Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martínez, S. R. White, J. S. Moore and N. R. Sottos, Nature, 2009, 459, 68–72, DOI:10.1038/nature07970
- M. Irie, Chem. Rev., 2000, 100, 1685–1716, DOI:10.1021/cr980069d
- D. Sud, T. B. Norsten and N. R. Branda, Angew. Chem., Int. Ed., 2005, 44, 2019–2021, DOI:10.1002/anie.200462538
- H. D. Samachetty and N. R. Branda, Chem. Commun., 2005, 2840–2842, 10.1039/B501779C
- T. van Leeuwen, T. C. Pijper, J. Areephong, B. L. Feringa, W. R. Browne and N. Katsonis, J. Mater. Chem., 2011, 21, 3142–3146, 10.1039/C0JM03626A
- S. J. van der Molen, J. Liao, T. Kudernac, J. S. Agustsson, L. Bernard, M. Calame, B. J. van Wees, B. L. Feringa and C. Schönenberger, Nano Lett., 2009, 1, 76–80, DOI:10.1021/nl802487j
- D. Vomasta, C. Högner, N. R. Branda and B. König, Angew. Chem., Int. Ed., 2008, 47, 7644–7647, DOI:10.1002/anie.200802242
- U. Al-Atar, R. Fernandes, B. Johnsen, D. Baillie and N. R. Branda, J. Am. Chem. Soc., 2009, 131, 15966–15967, DOI:10.1021/ja903070u
- M. Walko and B. L. Feringa, Chem. Commun., 2007, 1745–1747, 10.1039/b702264f
- K. Uchida, Y. Nakayama and M. Irie, Bull. Chem. Soc. Jpn., 1990, 63, 1311–1315, DOI:10.1246/bcsj.63.1311
- F. Stellacci, F. Toscano, M. C. Gallazzi and G. Zerbi, Synth. Met., 1999, 102, 979–980, DOI:10.1016/s0379-6779(98)01046-7
- A. J. Myles, Z. Zhang, G. Liu and N. R. Branda, Org. Lett., 2000, 2, 2749–2751, DOI:10.1021/ol006040p
- S. L. Gilat, S. H. Kawai and J.-M. Lehn, J. Chem. Soc., Chem. Commun., 1993, 1439–1442, 10.1039/c39930001439
- J. Areephong, T. Kudernac, J. J. D. de Jong, G. T. Carroll, D. Pantorott, J. Hjelm, W. R. Browne and B. L. Feringa, J. Am. Chem. Soc., 2008, 130, 12850–12851, DOI:10.1021/ja803714p
- F. Stellacci, C. Bertarelli, F. Toscano, M. C. Gallazzi, G. Zotti and G. Zerbi, Adv. Mater., 1999, 11, 292–295, DOI:10.1002/(SICI)1521-4095(199903)11:4<292::AID-ADMA292>3.0.CO;2-V
- T. Kawai, T. Kunitake and M. Irie, Chem. Lett., 1999, 905–906, DOI:10.1246/cl.1999.905
- M. Ranger, D. Rondeau and M. Leclerc, Macromolecules, 1997, 30, 7686–7691, DOI:10.1021/ma970920a
- Q. Luo, H. Cheng and H. Tian, Advance Article, Polym. Chem., 2011 10.1039/C1PY00167A
- C. Bertarelli, A. Bianco, V. Boffa, M. Mirenda, M. C. Gallazzi and G. Zerbi, Adv. Funct. Mater., 2004, 14, 1129–1133, DOI:10.1002/adfm.200304295
- H. W. Lee and E. Kim, J. Mater. Chem., 2006, 16, 1384–1389, 10.1039/b517175j
- H. Choi, H. Lee, Y. Kang, E. Kim, S. O. Kang and J. Ko, J. Org. Chem., 2005, 70, 8291–8297, DOI:10.1021/jo050710t
- T. Kawai, Y. Nakashima and M. Irie, Adv. Mater., 2005, 17, 309–314, DOI:10.1002/adma.200400191
- M. J. Marsella, Z.-Q. Wang and R. H. Mitchell, Org. Lett., 2000, 2, 2979–2982, DOI:10.1021/ol006293i
- R. H. Mitchell and V. Boekelheide, J. Am. Chem. Soc., 1974, 96, 1547–1557, DOI:10.1021/ja00812a045
- R. H. Mitchell, Eur. J. Org. Chem., 1999, 2695–2703, DOI:10.1002/(SICI)1099-0690(199911)1999:11<2695::AID-EJOC2695>3.0.CO;2-T
- G. P. Kittlesen, H. S. White and M. S. Wrighton, J. Am. Chem. Soc., 1984, 106, 7389–7396, DOI:10.1021/ja00336a016
- J. Yang and M.-K. Ng, Synthesis, 2006, 3075–3079, DOI:10.1055/s-2006-942529
- A. M. Fraind and J. D. Tovar, J. Phys. Chem. B, 2010, 114, 3104–3116, DOI:10.1021/jp9101459
| This journal is © The Royal Society of Chemistry 2011 |