Responsive hybrid block co-polymer conjugates of proteins–controlled architecture to modulate substrate specificity and solution behaviour†
Gökçen
Yaşayan
a,
Aram O.
Saeed
a,
Francisco
Fernández-Trillo
a,
Stephanie
Allen
a,
Martyn C.
Davies
a,
Abdulhakim
Jangher
e,
Alison
Paul
e,
Kristofer J.
Thurecht
b,
Stephen M.
King
c,
Ralf
Schweins
d,
Peter C.
Griffiths
e,
Johannes P.
Magnusson
*a and
Cameron
Alexander
*a
aThe School of Pharmacy, Boots Science Building, University of Nottingham, University Park, Nottingham, NG7 2RD, UK. E-mail: Johannes.magnusson@nottingham.ac.uk; cameron.alexander@nottingham.ac.uk; Fax: +44 (0) 115 951 5102; Tel: +44 (0) 846 7678
bAustralian Institute for Bioengineering and Nanotechnology (AIBN), Corner College and Cooper Roads (Building 75), The University of Queensland, Brisbane, Queensland 4072, Australia
cScience and Technology Facilities Council, Rutherford Appleton Laboratory, Harwell Science and Innovation Campus, Didcot, OX11 OQX, UK
dInstitut Laue-Langevin, 6 rue Jules Horowitz, 38000, GRENOBLE, France
eSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK
First published on 27th May 2011
Abstract
Responsive co-polymers based on polyethyleneglycol methacrylate (PEGMA) monomers have been grown by aqueous phase ATRP from a model protein, trypsin, to generate hybrid polymer-protein block conjugates. The conjugates (Hybrids I and II) both contained the same segment of grafted responsive co-polymer to afford a phase transition at 37 °C, Hybrid II however differed from Hybrid I by having a second block of hydrophilic pPEGMA monomer grown from the end of the responsive block. The resultant ‘diblock’ and ‘triblock’ hybrids were characterised in terms of their temperature-dependent behaviour in solution by dynamic light scattering, small-angle neutron scattering and pulsed-gradient spin-echo NMR, and their structures at surfaces examined by aqueous phase atomic force microscopy and cryo transmission electron microscopy. These data showed that Hybrids I and II differed in their solution behaviour with temperature, dependent on the arrangement of their grafted polymer blocks. Hybrid I self-assembled into higher-order structures above 37 °C before precipitating reversibly, whereas Hybrid II remained essentially constant in size across a similar temperature range even when its attached intermediate polymer block underwent a phase transition. The differences in polymer-protein hybrid behaviour were also manifest in enzyme activity assays with temperature-dependent hydrolysis of both peptide and protein substrates varying with hybrid architecture. Overall the data show that it is possible to grow responsive polymer-protein block co-polymers of varied structures, architectures and solution behaviour and that these can be used to control bioconjugate activity.
Introduction
Proteins and enzymes are increasingly used in biotechnology and medicine, but the utility of many is hampered by short half-life profiles in the bloodstream and limited stability in vitro.1 These shortcomings can be addressed by engineering selective changes in amino acid sequences to create more stable derivatives, or by creating a protective/stabilising layer for the protein by attachment of polymers. In principle, the protective shell could be any material that is hydrophilic, stable and sterically shielding, but in practice poly(ethylene glycol) (PEG)-based polymers have been the most investigated.The popularity of PEG polymers for bioconjugation arises from their inert chemistry once attached and their favourable toxicology profile, which has led to FDA approval.2 However, in certain applications the inertness of PEG polymers can be disadvantageous, such as where strong cellular/membrane interactions are required, or if recycling of protein is necessary for a bioprocessing use. Moreover, attachment of non-functionalised PEG chains normally generates a permanent change in the activity of the protein, which cannot be manipulated to suit the inherently dynamic biological environment. It is thus highly desirable to devise protein conjugation strategies that enable the activity of the protein to be modulated in a controllable way.
Conjugation of responsive or ‘smart’ polymers offer a means by which this functional or behavioral change in a protein can be introduced and then controlled by an external stimulus. In general, responsive polymers exhibit a non-linear change in their properties following a stimulus, such as change in temperature, pH or via radiation.3 Polymers of this type have been used in the bioconjugation context to aid in the recovery of proteins,4 improve their stability,5 or increase accumulation in a target tissue.6 To date, the most extensively studied “smart” polymer is poly N-isopropylacrylamide (PNIPAm)7 which exhibits a lower critical solution temperature (LCST) at 32 °C. The effect of PNIPAm on the activity of enzymes, and its application in the recovery of proteins have been widely reported.4,8 Unfortunately, there are some concerns over potential PNIPAm cytotoxicity,9 and as a consequence, “smart” poly(ethylene glycol methacrylate) (PEGMA) based copolymers10 have been proposed as potential biocompatible alternatives.11 The similarity of the PEGMA based polymers to PEG makes them promising candidates for biomedical applications.
Of particular interest is the possibility of using responsive PEGMA-type polymers as multi-faceted protein activity modifiers, wherein the variable conformations displayed by the polymer chains as they respond to stimuli invoke different functional properties of the protein. The additional advantage of PEGMA materials is their accessibility in a range of architectures through controlled polymerisation techniques. In previous papers we have described a fully aqueous route to responsive PEGMA materials,12,13 and a number of PEGMA-protein conjugates have now been prepared.14–18
Here we show how PEGMA based polymers can be used to form responsive copolymer-protein conjugates by growing directly from the functionalised protein and, importantly, that the sequence and architectures of the attached co-polymers have a critical effect on the structures of the conjugates and on the activity of the conjugated protein. Thermoresponsive PNIPAm polymers have been grown from proteins previously19–21 but to our knowledge growing a thermoresponsive PEGMA polymer from a biomacromolecule has not previously been reported. Trypsin was selected as the model protein due to its well known proteolytic activity and its importance in biotechnology applications.22 Activity data for trypsin-polymer conjugates have also been reported previously,23–27 and very recently a responsive PEGMA-trypsin polymer was described.15 Through an extension of the fully aqueous atom transfer radical polymerization (ATRP) route, we have been able to prepare different polymer-protein architectures from a common trypsin ancestor, and have evaluated the solution behaviour and activity of these hybrids. We demonstrate that the architectures of the co-polymers and the structures of the two trypsin hybrid conjugates (Hybrids I and II, Scheme 1) change markedly following temperature stimuli across the phase transition temperatures, and that these structures in turn can be used to modulate the properties of the protein polymer hybrids in terms of stability and reactivity.
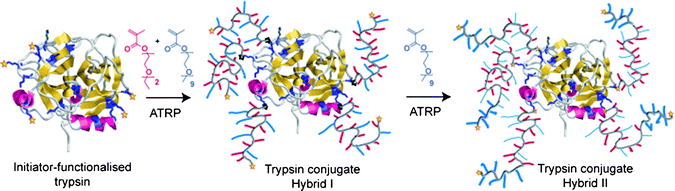 | ||
| Scheme 1 Design criteria of trypsin conjugates with a statistical responsive co-polymer hybrid (trypsin–Hybrid I) and a triblock (trypsin Hybrid II). | ||
Materials and methods
All solvents and reagents were of analytical or HPLC grade and purchased from Sigma or Fisher Scientific unless otherwise stated. Deuterated solvents were purchased from Sigma.Polyethylene glycol ethyl ether methacrylate (PEGMA-EE 246, Mn 246), and polyethylene glycol methyl ether methacrylate (PEGMA-ME 475, Mn 475) were purchased from Sigma Aldrich and purified before use by passing through a column filled with neutral alumina. N-α-Benzoyl-DL-arginine p-nitroanilide hydrochloride (BAPNA) (>98%), α-bromoisobutyryl bromide (98%), benzamidine hydrochloride (99%), casein (sodium salt), copper(II) bromide (CuBr2, 99%), L-ascorbic acid (99%), N,N′-disuccinimidyl carbonate (DSC) (>95%) tetraethylene glycol (99%), triethylamine (99.5%), and trypsin from porcine pancreas were used as received from Sigma Aldrich. Phosphate Buffer Saline (PBS) was used as received from Fisher Scientific. 1X PBS used is equivalent to a concentration of 137 mM NaCl, 2.7 mM KCl and 10 mM phosphate buffer. Water used for the polymerisations was of ELGA grade. Tris(2-pyridyl) methylamine (TPMA) was prepared as described elsewhere.28 Dialysis membrane (MWCO 6-8000, regenerated cellulose) was used as received from Spectrapor. Centrifugation was carried out using a Centaur II centrifuge.
For AFM studies, mica discs and specimen discs were purchased from Agar Scientific (Stansted, UK), SNL-10 (Sharp Nitride Lever-10) AFM probes were purchased from Bruker (Cambridge, UK). Syringe filters (20 nm, Anotop 10) were purchased from Whatman (Kent, UK) and 0.2 μm syringe filters were purchased from Interlab (Wellington, New Zealand).
Measurements and analysis
Gel permeation chromatography (GPC)
Molecular weights and molecular weight distributions were determined using a Varian/Polymer Laboratories GPC-50 instrument with triple detection (RI, viscometry and MALLS). Chromatograms were run at 40 °C using chloroform (CHCl3) as eluent with a flowrate of 1 ml min−1. The columns used were Resipore Mixed-D, detection was performed by a Refractive Index detector (RI). The machine was calibrated with linear polystyrene standards.Polymers were cleaved specifically by adapting a method from Jaquemard et al.29 The protein polymer hybrid (20 mg), 10 mL of anhydrous THF and 200 μL of tetra-n-butylammonium fluoride (TBAF) were placed in a round bottom flask with a condenser and the mixture was refluxed for 6 h. The THF was evaporated and the polymer extracted into chloroform and analysed by GPC.
SDS PAGE
SDS-PAGE gel electrophoresis was carried out at 100 mV using 8% acrylamide running gel and 4% stacking gel prepared by the standard method (non denaturating conditions) and visualised by Coomassie staining.30The gels were additionally stained to detect PEG using the barium/iodine method.31
DLS (Dynamic light scattering)
Hydrodynamic radii of the protein polymer hybrids in solution were measured via scattered light recorded at a 90° angle to incident radiation in a Viscotek 802 dynamic light scattering (DLS) instrument equipped with a 50 mW internal laser operating at a wavelength of 830 nm. From standard auto correlation functions, measured diffusion coefficients were related to particle hydrodynamic radius via the Stokes–Einstein equation| RH = kT/6πηD |
Measurements quoted are the averages of triplicate samples of six replicates with at least 10 readings of particle size recorded at each temperature. Radii quoted are averages for samples where >75% of the scattered light in terms of particle masses was from polymers within the size range quoted unless otherwise stated.
Transmission Electron Microscopy (TEM)
Samples were prepared on carbon/formvar grids which were made hydrophilic by argon plasma cleaning (in a Fischione Model 1020 Plasma Cleaner) prior to use. Samples were spotted onto the grid with a Cryo-plunge (CP3 Gatan Inc) and were instantaneously frozen using liquid ethane, creating a frozen hydrated sample in a thin film of vitreous ice.Solutions of each hybrid (1 mg mL−1) in 0.05 M Tris and 20 mM CaCl2 buffer were prepared, the solutions were filtered through a 200 nm syringe filter. Samples below the cloud point temperature were spotted onto grids without any further treatment. Samples above the cloud point temperature were heated for 5 min at 70 °C prior to the solution spotting process. In these cases the appropriate solution (5 μL) was placed directly on the Cryo-plunge before application on the chilled grid. Cryo-TEM spectroscopy was carried out on a JEOL Ltd JEM-2100F microscope with a Gatan 914 Cryo-tomography holder. Images were taken with a Gatan Orius camera.
Atomic Force Microscopy (AFM)
Topography images and particle analysis of Hybrid I and Hybrid II were acquired in liquid with AFM (MultiMode Scanning Probe Station with Nanoscope IIIa controller (Bruker, Santa Barbara, CA) operating tapping mode. Images were acquired using an E-scanner, at scan rates between 5–8 Hz.AFM height images of trypsin were obtained in liquid at room temperature using a Multimode 8 Scanning Probe Microscopy station, operating in PeakForce Tapping™ mode. This imaging mode permitted greater control of probe-sample contact force and facilitated imaging of these samples. Images were acquired using an E-scanner, at scan rates between 1–2 Hz.
AFM studies on the hybrids were carried out at different temperatures using an external heating stage (Nanoscope, Bruker, Santa Barbara, CA). The samples were injected onto freshly cleaved mica at 30 °C and the temperature increased to 40 °C; finally the temperature was reduced to 30 °C. These experiments were designed to assess the aggregation behaviour of the protein-polymer hybrids and determine whether the aggregation was reversible.
For AFM experiments all protein and protein-polymer conjugate solutions were prepared at a concentration of 10 μg ml−1 with 0.05 M Tris buffer, containing 20 mM CaCl2 (pH 8.2) and filtered with a 20nm syringe filter. Image data were analysed using NanoScope Analysis software (Version 1.20 (Bruker)).
MALDI TOF (Matrix-assisted laser desorption/ionisation–time of flight)
MALDI TOF analysis was carried out on a Bruker MALDI TOF Ultraflex II machine.Samples were prepared as described elsewhere.32 Briefly an aqueous solution of the conjugate (1.0 mg mL−1) was mixed with an equal volume of matrix material (8 mg sinapic acid, 0.5 mL water and 0.5 mL MeCN). An aliquot (2 μL) of the resulting mixture was spotted on to a plate target and allowed to dry. The level of conjugation was determined by comparing the molecular weight of conjugate to native trypsin.
Pulsed-Gradient Spin-Echo NMR (PGSE-NMR)
Measurements were conducted on a Bruker AMX360 NMR spectrometer using a stimulated echo-sequence, as described elsewhere.33 This configuration uses a 5 mm diffusion probe (Cryomagnet Systems, Indianapolis) and a Bruker gradient (GRASP) spectroscopy accessory unit to deliver trapezoidal gradient pulses.The self-diffusion coefficient Ds was extracted by fitting the integrals for a given peak to eqn (1);
| A(δ,G,Δ) = A0exp(−kDs) | (1) |
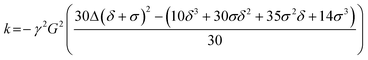 | (2) |
Small-Angle Neutron Scattering (SANS)
Small-angle neutron scattering (SANS) measurements were performed on two instruments-the fixed-geometry, time-of-flight LOQ diffractometer (ISIS Spallation Neutron Source, Oxfordshire UK) and the fixed-wavelength D11 diffractometer (ILL, Grenoble, France). On LOQ, neutron wavelengths spanning 2.2 to 10 Å were used to access a Q range (Q = 4πsin(θ/2)/λ) of approximately 0.008 to 0.25 Å−1 (25 Hz), with a fixed sample-detector distance of 4.1m. On D11, the wavelength was set at 8 Å, and three sample-detector distances were employed to span a comparable Q range. On both cameras, the samples were contained in 2 mm path length, UV-spectrophotometer grade, quartz cuvettes (Hellma) and mounted in aluminium holders on top of an enclosed, computer-controlled, sample chamber. Sample volumes were approximately 0.4 cm3. Temperature control was achieved through the use of a thermostated circulating bath pumping fluid through the base of the sample chamber. Under these conditions a temperature stability of better than ± 0.5 °C can be achieved. Experimental measuring times were approximately 40 min.All scattering data were (a) normalised for the sample transmission, (b) background corrected using a quartz cell filled with D2O (this also removes the inherent instrumental background arising from vacuum windows, etc.) and (c) corrected for the linearity and efficiency of the detector response using the instrument-specific software package. The data were put onto an absolute scale by reference to the scattering from a partially deuterated polystyrene blend (LOQ) or 1 mm water (D22).
BCA (Bicinchoninic acid) protein assay
Protein content quantification of protein polymer hybrids was assessed using the BCA assay of Smith et al.34 In a typical assay, solutions (3.0 mg mL−1) of the protein polymer hybrids were incubated with a BCA/copper solution and the absorbance read at 562 nm. This absorbance was compared to a calibration from a bovine serum albumin standard.Cloud-point and Lower Critical Solution Temperature (LCST) determinations
A protein polymer stock solution (3.0 mg mL−1) was prepared using double distilled water (DDW) and the appropriate salts. The UV absorption for each sample was measured at a wavelength of 550.0 nm over a temperature range of 20.0 °C–65.0 °C. The temperature was controlled and measured using a peltier plate heating system (Beckman) and was increased at a rate of 0.5 °C min−1. We considered the cloud point to be the onset of a sharp increase in UV absorption at 550 nm in accordance with prior studies.35 A Beckman Coultier DU 800 UV spectrophotometer with a thermostat was used for activity assays and cloud point measurements.Trypsin activity assay and enzyme kinetics using BAPNA (N-α-Benzoyl-DL-arginine 4-nitroanilide hydrochloride)
The activity of the trypsin hybrids towards a small molecule substrate was estimated according to the method of Erlanger et al.36 A fresh solution of BAPNA was made by dissolving 40 mg in 2.5 mL of DMSO and diluting to 50 mL with 0.05 M Tris HCl/20 mM CaCl2 buffer—pH 8.2. 280 μL of BAPNA solution was used for each assay. The solution was equilibrated at appropriate temperature for 10 min, then 20 μL of trypsin aliquots (160 μg mL−1 eq. of native trypsin) were added, initial absorbance change was recorded at 410 nm. Relative activity of the hybrids was estimated by comparing the change in absorbance to that of native trypsin at 22 °C.Michealis Menten plots were derived for the polymer hybrids and native protein at 26 °C and 40 °C respectively. 185 μL of BAPNA solution (at various concentrations) were used for each assay, the solution was incubated on a UV spectrometer with a temperature control for 10 min before adding 15 μL of protein solution (160 μg mL−1 eq. of native trypsin) to the cuvette. The change in absorbance at 410 nm was read immediately for each of the concentrations. All experiments were carried out in triplicate.
Trypsin protease activity towards casein
The activity of the hybrids against a macromolecular substrate was estimated according to the method of Singh and Krikorian37 A 2.5% solution of casein in Tris buffer (0.05 M Tris HCl, 20 mM CaCl2 pH 8.2) was prepared. Casein solution (1.0 mL) and Tris buffer (350 μL of 0.05 M Tris HCl; 20 mM CaCl2 pH 8.2) were used for each assay. The solution was equilibrated at the appropriate temperature for 10 min after which trypsin aliquots (100 μL of 80 μg mL−1 of trypsin eq.) were added and incubated for 20 min. The reaction was stopped by adding 10% trichloroacetic acid (2.0 mL) to the solution, the mixture was centrifuged at 4000 rpm for 30 min and the clear supernatant collected. The absorbance of the solution was recorded at 280 nm and then compared to absorbance of a native trypsin solution to determine the relative activity. All experiments were carried out in triplicate.Trypsin thermal stability
Native trypsin and hybrids solutions were incubated at room temperature and 37 °C over 2 days (160 μg mL−1 eq. of native trypsin) in 1X PBS buffer at pH 7.4. The residual activity was measured using the BAPNA assay by incubating 185 μL of BAPNA solution with 15 μL of protein solution for 10 min at room temperature. The initial change in absorbance was measured at 410 nm and compared to that of the protein solutions prior to heating.Modification of trypsin: conjugation of ATRP initiator functionality trypsin
Linker synthesis was carried out as previously reported.14 Trypsin (760 mg, 0.034 mmol) and benzamidine hydrochloride (48 mg, 0.30 mmol) were dissolved in 76 mL of 100 mM PBS and the pH was adjusted to 7.5. The amine reactive triethylene glycol ATRP initiator (400 mg, 0.82 mmol) was dissolved in 1.5 mL of DMSO and added dropwise, the reaction was left to react for 90 min. The mixture was dialysed with a cellulose membrane MWCO 6-8000 for 2 days at 4 °C, centrifuged at 4000 rpm, the supernatant collected and lyophilised. The total amount of modified trypsin was 760 mg.Responsive co-polymers to match those grafted from Hybrid I and Hybrid II, i.e. a (PEGMA-EE-246)85-stat-PEGMA-ME-475)15 (Co-Polymer 1) and a [(PEGMA-EE-246)85-stat-PEGMA-ME-475)15]92-graft-(PEGMA-ME-475)50 (Co-Polymer 2) were prepared by our previous methods.12
Results
The pre-requisite for preparing responsive co-polymer conjugates with varying architectures required a method to grow statistical and block materials from the protein of choice, in this case, trypsin. Our prior routes to “smart” PEGMA copolymers12 utilised polyethylene glycol ethyl ether (PEGMA-EE-246; Mn 246) and polyethylene glycol methyl ether (PEGMA-ME-475; Mn 475) methacrylates, with varying monomer feed and the AGET ATRP method38 to produce co-polymers under fully aqueous conditions. These afforded polymers with LCST values across a wide temperature range. Accordingly, we needed ATRP initiator sites on the protein, and thus synthesised a heterobifunctional linker with an amino-reactive terminus and a radical generating center (Scheme 2). The linker was synthesised from a tetraethylene glycol (TEG) precursor via a published route.14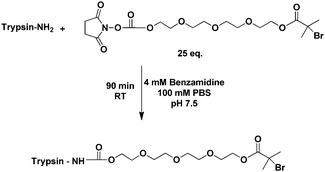 | ||
| Scheme 2 Conjugation of initiator-functionalised trypsin. | ||
The initiator was conjugated to accessible lysine residues on the protein through a succinimidyl succinate reactive group, in phosphate buffered saline (PBS) with an excess of the ATRP initiator and benzamidine hydrochloride present. This generated the ‘core’ functionalised trypsin (Scheme 2 and Fig. 1a) used in subsequent polymerisation experiments. Benzamidine was used to protect the active sites of trypsin from potential conjugations and to prevent autocatalysis since it is a known inhibitor of the enzyme.39
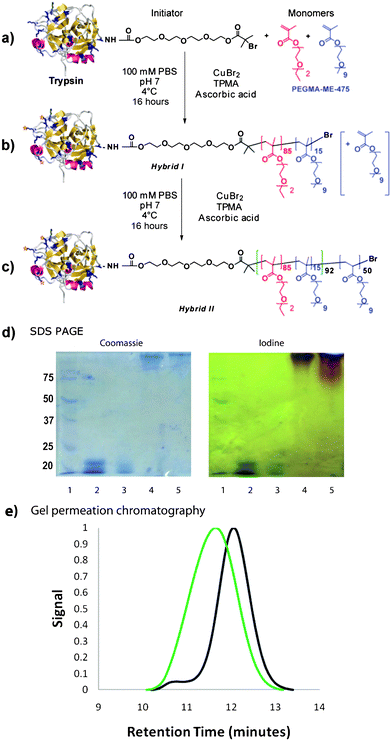 | ||
| Fig. 1 Synthetic scheme for initiator-functionalised trypsin (a), Hybrid I (b) and Hybrid II (c). SDS PAGE (d) for Hybrid I and II with stains for protein (Coomassie) and PEG (iodine) indicate success of polymer growth from trypsin (Lane 1: Marker; Lane 2: Native Trypsin; Lane 3: Initiator-functionalised Trypsin; Lane 4: Hybrid I, Lane 5: Hybrid II). e) Gel permeation traces of polymer after cleavage from protein. (Black trace–Hybrid I, Green Trace–Hybrid II). | ||
The polymerisation from trypsin was carried out at low temperature (4 °C) under aqueous conditions. A polymer with a targeted LCST of 37 °C was initially grown from the functionalised trypsin protein to create co-polymer trypsin Hybrid I (Table 1, Fig. 1b). The purified polymer conjugate was then split into portions, of which one was used to initiate a second polymerisation with PEGMA-ME-475 to create the triblock trypsin Hybrid II. (Table 1, Fig. 1c).
| Hybrid | Initiatorsa | M n | M w/Mnb | LCSTc | BCAd |
|---|---|---|---|---|---|
| a Average number of initiating sites as determined with MALDI TOF. b From GPC—length of each polymer chain. c Cloud point measured by UV absorption of 3 mg mL−1 solutions in 1X PBS at 550 nm. d Protein content determined by bicinchoninic acid assay (BCA). | |||||
| I | 5.1 | 28300 | 1.19 | 36 °C | 14.82% |
| II | 5.1 | 45606 | 1.53 | 36 °C | 9.14% |
Electrophoresis (SDS PAGE, Fig. 1d) revealed that all of the activated trypsin had polymerised to form Hybrid Ii.e. no native or initator-functionalised trypsin was observed in the SDS PAGE gel after polymerisation. An additional low molecular weight band was observed in the gel for the native trypsin and to a less extent for the initiator functionalised trypsin, which stems from its autocatalysis. This was not observed for the polymerised trypsin as the polymer chains decreased autocatalytic breakdown, most likely due to increased steric hindrance. Neither hybrid penetrated the gel to a great extent due to their bulkiness. Characterisation (Fig. 1e) of the polymer species grown from the proteins was achieved by treatment of the conjugates with tetrabutylammonium fluoride to cleave the polymer-protein links. GPC analysis indicated that the polymer cleaved from Hybrid I was mostly of narrow polydispersity although a small tail was observed towards the high molecular weight range; the calculated polydispersity for the main peak was 1.19. The polymers from Hybrid II exhibited broader molecular weight distribution (PDI 1.53) than their precursors but the peak observed was symmetrical. Comparison of the two chromatographs clearly showed that Hybrid I had efficiently initiated the polymerisation of PEGMA-ME-475 to form Hybrid II.
The solution properties of Hybrids I and II were examined by means of dynamic light scattering (DLS, ESI†) and UV turbidity as a function of temperature. Both conjugates were shown to exhibit temperature-dependent solution behaviour, but the specific effects were different for each polymer, and strongly dependent on the ionic environment of the solution. (Fig. 2)
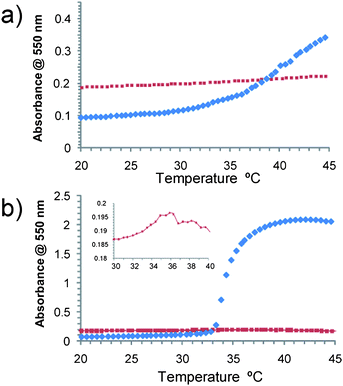 | ||
| Fig. 2 Temperature-turbidity curves in 1X PBS (top) and Tris buffer (0.05 M + 20 mM CaCl2 pH 8.2, bottom). Trypsin Hybrid I (blue trace). Trypsin Hybrid II (red trace). (Expanded area—Hybrid II in Tris buffer). | ||
In isotonic phosphate-buffered saline (PBS) solution, both hybrids exhibited increases in turbidity at raised temperature, although the increase was much smaller for Hybrid II than Hybrid I. In Tris buffer, Hybrid I showed a large increase in turbidity above the LCST, indicating aggregation or assembly to larger objects. Conversely, Hybrid II exhibited a slight decrease in turbidity at the onset of LCST, implying that above the LCST the aggregation state or size of objects present in solution decreased. DLS experiments (ESI†) recorded in Tris buffer showed that in all cases, mixed populations of species were present in suspensions, indicating some heterogeneity in the conjugates across the temperature ranges. Particle size distributions based on scattered light intensity were dominated by the larger species, but estimated sizes based on number distributions suggested that for both Hybrid I and II in Tris buffer, non-aggregated particles were the majority species. For Hybrid II in Tris buffer, the most numerous particles were those with an average RH of around 5–7 nm. The size was not greatly affected by the onset of LCST but seemed to decrease slightly (in both intensity and number distributions), which was in accordance with the decreased turbidity observed previously. In the same buffer, the most numerous Hybrid I particles exhibited an average RH of 5 nm below the LCST but a large increase to 150 nm above the LCST (see ESI†). The different solution behaviour between the two buffers stems from their different salting out/salting in properties, we have previously demonstrated that the thermoresponsive behaviour of PEGMA is significantly affected by the ionic environment.12 In order to evaluate the temperature-dependent behaviour further, cryo-TEM and AFM analysis were carried out on the hybrids in Tris buffer to acquire information on the morphology of the conjugates.
Cryo-TEM analysis (Fig. 3) suggested that Hybrid I formed mainly discrete spherical objects of 70–80 nm diameter below the LCST. When the temperature was raised above the LCST larger aggregates were formed. The aggregates of Hybrid I themselves appeared from the cryo-TEM images to be composed of smaller particles which were around 30–40 nm in diameter (Fig. 3b, inset). In comparison, for Hybrid II the onset of LCST did not markedly affect the observed size, which remained around 20–50 nm below and above the LCST. For Hybrid II also the particles on close inspection appeared to be conglomerates of smaller particles, of ∼10–20 nm diameter (Fig. 3d, inset).
 | ||
| Fig. 3 Cryo-TEM imaging of trypsin conjugates in 0.05 M Tris and 20 mM CaCl2 buffer. a) Hybrid I flash-frozen from solution at temperatures below the LCST, b) Hybrid I flash-frozen from solution at temperatures above the LCST, c) Hybrid II frozen from below the LCST, d) Hybrid II frozen from above the LCST (scale bars 0.2 μm). Insets to (b) and (d) show particles at higher magnification. | ||
It was not possible to discern whether the sizes and morphologies of the particles and aggregates in the images were inherent or influenced by dehydration/flash-freezing artefacts by TEM alone. Accordingly, we investigated the morphologies of the protein-polymer conjugates by AFM of particles in aqueous suspension. The images obtained showed that under all conditions the conjugates were approximately spherical, although there was a size range of particles across all the samples (Fig. 4).
 | ||
| Fig. 4 Selected AFM topography images of a) Hybrid I and b) Hybrid II. The system was heated to 30 °C (left hand panel), then to 40 °C (middle panel) before being cooled to 30 °C (right hand panel). Scale bars are 100 nm. Vertical scale is 8 nm for Hybrid I at 40 °C and 2 nm for the other images. | ||
To probe their self-assembly behaviour, a heating stage was utilised, in situ, and samples were imaged at 30 °C before increasing the temperature to 40 °C. Following imaging at this increased temperature, samples were cooled to 30 °C and then imaged again. Samples were left for at least 30 min following temperature increases/decreases, to allow samples to equilibrate prior to resuming imaging.
Grain size analysis of the features observed in AFM at each temperature showed that the mean diameter of the Hybrid II remained between 7–11 nm at all temperatures, while the mean diameter of Hybrid I increased from 7 nm (at 30 °C) to ∼ 24 nm (at 40 °C), with the maximum size at this temperature of 58 nm (Table 2). The mean particle size of Hybrid I returned to ∼ 11 nm when the system was cooled down to 30 °C, possibly reflecting that full dissociation of the aggregates was hindered by the presence of underlying mica surface.
| Total particle count | Mean diameter (nm) | Minimum diameter (nm) | Maximum diameter (nm) | |
|---|---|---|---|---|
| Trypsin | 87 | 4.3 | 3.3 | 8.1 |
| Hybrid I at 30 °C (i) | 99 | 6.8 | 3.7 | 14.2 |
| Hybrid I at 40 °C (ii) | 101 | 23.7 | 10.3 | 58.3 |
| Hybrid I at 30 °C (iii) | 8 | 11.2 | 7.6 | 15,8 |
| Hybrid II at 30 °C (i) | 98 | 7.4 | 3.7 | 14.9 |
| Hybrid II at 40 °C(ii) | 122 | 7 | 3.4 | 17.5 |
| Hybrid I at 30 °C (iii) | 50 | 10.6 | 3.4 | 19.1 |
Further characterisation of the solution conformation of the hybrids, and their PEGMA analogues, was carried out by pulsed-gradient spin-echo NMR (PGSE-NMR) and small-angle neutron scattering (SANS). The self-diffusion coefficients measured by PGSE-NMR were recast in terms of the corresponding hydrodynamic radii and the temperature dependence of this estimate of the solution conformations are presented in the ESI.† From this data trypsin was shown not to display any marked change in solution conformation over the temperature range 20–35 °C. The PEGMA polymers—here used as models for the thermoresponsive grafts grown from the trypsin core—did show the expected monotonic decrease in hydrodynamic radii over the same temperature range. However, the NMR diffusion experiments were performed at temperatures below the LCSTs owing to the difficulties in interpreting signal broadening of the polymers alone as they precipitated at the LCST.
For the polymer-trypsin conjugates, i.e. Hybrid I and II, rather different temperature responses were observed compared to each other and compared to the native enzyme and the non-conjugated polymers. Hybrid I showed a monotonic decrease in hydrodynamic radius with increasing temperature, similar to that observed for the polymer model itself. The radius of the conjugate was always greater than that of the free PEGMA polymer and the trypsin. Hybrid II on the other hand displayed a different temperature profile, showing a pronounced decrease in size around 28 °C, with the size at the higher temperature approaching that of the trypsin core. The collapse of the triblock grafts thus appeared to give rise to a changed architecture of the conjugate relative to polymer collapse in the diblock conjugate, with both qualitative and quantitative agreement with the behaviour observed in the AFM studies.
Additional insight was gained by considering the solution neutron scattering. Data are presented in two formats—as a conventional (raw data) plot (I(Q) vs. Q) in the ESI,† and in the form of a Kratky plot (Q2I(Q) vs. Q, which emphasises the departure from Gaussian statistics of the polymer conformation) which yielded information on the differences in the two conjugate structures and their variation with temperature. The conventional plot indicated that with increasing temperature, Hybrid I (diblock) and Hybrid II (triblock) polymers showed an increase in scattering. At low temperatures the monotonically decaying data were well-described by a simple polydisperse Gaussian coil model, with radii of gyration of few nanometres, consistent with the AFM and NMR estimates of the solution conformation. At higher temperatures, points of inflexion were evident in these exemplar data, indicating that the structures present possessed delineated or phase separated regions with different scattering length densities, i.e. core–shell structures. Indeed, such a model fitted the data well, with core radii of a few nanometres and a slightly thicker shell, although these quantities should not be over interpreted in the absence of other confirmatory techniques.
The Kratky representation emphasised these differences rather more markedly. The scattering from a typical Gaussian coil, which varies as I(Q) ∼ Q−2 would be expected to increase to a plateau, attaining a horizontal asymptote. A compact or globular structure would exhibit a maximum at a Q value corresponding to some characteristic length scale, e.g. a radius of gyration or correlation length.
As may be seen and as expanded in the ESI,† the PEGMA diblock (Fig. 5a) and triblock (Fig. 5b) polymers at low temperature exhibited the behaviour expected for a Gaussian coil, but showed pronounced peaks as the temperature approached that at which the polymer phase separated. No macroscopic phase separation was observed in these samples. Again, Hybrid I and II were easily distinguished by their temperature profiles—Hybrid I (Fig. 5c) showed a pronounced peak at Q = 0.025 Å−1, corresponding to R = 25 nm, whereas Hybrid II (Fig. 5d) showed only a change in slope at this Q value. This suggests that Hybrid I possessed a much more compact structure, or associated into a higher order structure with a characteristic dimension somewhat larger than the single bioconjugate molecule precursor, but Hybrid II did not self-assemble in the same way.
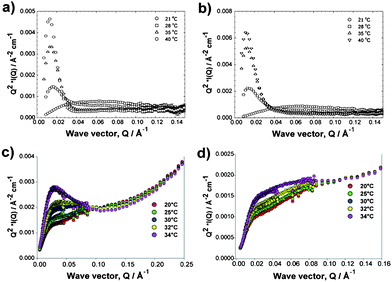 | ||
| Fig. 5 Temperature dependence of the small-angle neutron scattering from the co-polymer analogues 1 and 2 (a,b) and their trypsin hybrids (bottom) in PBS/D2O for Hybrid I (c) and Hybrid II (d). | ||
Having shown the variation of polymer architecture on the assembly of Hybrid I and II, we investigated the effect on trypsin stability and activity. Initial experiments focused on trypsin-mediated hydrolysis of N-α-benzoyl-DL-arginine p-nitroanilide hydrochloride (BAPNA). Assays were carried out using native trypsin and the Hybrid I and II conjugates with different concentration of BAPNA and at temperatures below and above the LCST (Fig. 6). The data showed that modification of the protein increased stability over time towards BAPNA hydrolysis (Fig. 6a) but decreased the turnover rate of the enzyme and its affinity for the peptide substrate (Fig. 6b and c). There was however a significant difference between the two hybrids with temperature change. Below the LCST of the attached polymers (Fig. 6b) both hybrids exhibited similar turnover rate and affinity for the low molar mass BAPNA substrate. Above the LCST (Fig. 6c), Hybrid I exhibited higher turnover than Hybrid II but significantly lower affinity for the substrate.
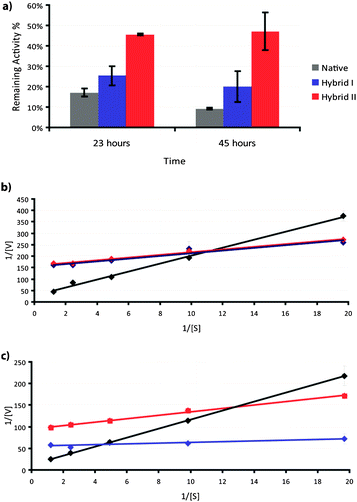 | ||
| Fig. 6 Enzyme kinetics and thermal stability of hybrids. a) Thermal stability of hybrids at 37 °C in 1X PBS pH 7.4 as a function of time. (Native—grey trace, Hybrid I—blue trace, Hybrid II—red trace). Lineweaver–Burk plots for BAPNA substrate below LCST b) 26 °C and above LCST c) 40 °C. (Native—black trace, Hybrid I—blue trace, Hybrid II—red trace). | ||
In assays to probe whether variations in polymer-enzyme conjugate self-assembly were responsible for the differences in Hybrid I and Hybrid II, we examined the activities of the conjugates towards substrates of different size. The activities of the hybrids were studied, again using the model peptide BAPNA, but compared with a protein (casein) to assess hydrolysis of a higher molar mass substrate. The activities were mapped across a range of temperatures around the LCST of the conjugates and compared to the activity of native trypsin (Fig. 7).
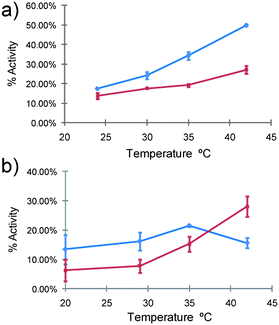 | ||
| Fig. 7 Reactivity of hybrids towards different substrates as a function of temperature. a) BAPNA assay (small substrate) b) Casein assay (large substrate). Hybrid I (blue trace), Hybrid II (red trace). The activity of native trypsin at 22 °C (BAPNA) and 20 °C (Casein) was set in each case as the reference activity (i.e. 100% activity at the start time). | ||
In terms of overall activity, both hybrids exhibited a reduced ability to hydrolyse the substrates, as expected owing to increased steric hindrance on the surface of the conjugates and their larger hydrodynamic volume. However, in accord with the kinetics assays, a notable difference in activity between the two hybrids was apparent. When compared in terms of activity against the small peptide substrate, Hybrid I caused a greater extent of hydrolysis than Hybrid II across the whole temperature range. For the casein substrate the activity of Hybrid I was initially higher than Hybrid II, but as the temperature reached the LCST, the activity reached a maximum and then decreased to an extent lower than the activity of Hybrid II. Casein is known to form nanoclusters and higher-order structures (“casein micelles”) in solution,40 and thus the different activities of the hybrids towards casein hydrolysis were likely to be a function of the different self-assembly modes of the conjugates at the varying temperatures.
Discussion
The starting point for this investigation was the desire to produce ‘active’ bioconjugates with functionalities that could be controlled by a responsive synthetic polymer. We also aimed to do this using chemistry that would be amenable to a variety of delicate biomolecules, and which would allow a range of polymer architectures to be assembled easily. Although there have been a large number of reports of responsive polymer-protein conjugates, the full possibilities for preparing these conjugates in multicomponent architectures have only recently begun to be explored.41–45 In addition, while the richness of block co-polymer structures has been opened up enormously by controlled radical techniques, the ability to synthesise as well as characterise polymer-biopolymer conjugates as block co-polymers is relatively new. We therefore set out to prepare responsive polymer-protein conjugates as hybrid block co-polymers, and to compare a ‘conventional’ polymer-protein “di-block” conjugate with a novel “tri-block” architecture consisting of a responsive polymer in between an enzyme block and a non-responsive hydrophilic block.46 The strategy behind the work was informed by the contrasting practical advantages and disadvantages of protein conjugation. In general, attachment of polymers such as PEG improves the stability of proteins to non-specific absorption and/or degradation in the bloodstream by creating a steric shield on the protein's surface. However, polymer conjugation can at the same time generally have detrimental effects on the activity of the protein by decreasing the accessibility towards substrates and receptors.47 The effect is dependent on the nature and length of both the linker and polymer, and the extent of conjugation.48,49 Deactivation of the protein can be alleviated by attachment of polymers to sites away from the catalytic centres, but this is not possible in all cases even though a number of very elegant strategies have been developed for site-specific conjugation.38,50,51 A possible solution to this problem is the use of a polymer that can shield the protein under one set of conditions, but collapse to expose the active site under another set of conditions. Pioneering work by the Hoffman group established this concept, using thermoresponsive poly(NIPAm).52,53 The drawback to this procedure is that collapse of a thermosensitive polymer attached to a protein in effect leads to the formation of an amphiphilic block co-polymer. In turn this may exhibit self-assembly into more complex architectures that confound predictions of protein activity.Accordingly, we set out to grow responsive polymers from an active protein, with the simultaneous aim of exploring whether “di- and tri-block” architectures could be prepared from a protein under fully aqueous conditions, and also whether these architectures could be used to control enzyme activity in different ways. A related goal was to establish whether a triblock responsive polymer-protein architecture could be used to confer ‘switchable’ behaviour but without causing phase separation of the conjugate as can occur with the more simple diblock polymer-protein structures. Finally, we aimed to assess whether enzyme stability and activity altered with conjugate architecture in comparison to the native enzyme.
The first steps in the protocol involved attachment of initiator functionality, in the presence of benzamidine to inhibit proteolysis during the coupling step. As shown in Scheme 2, generation of an α,ω-functional succinimidyl carbonate ATRP initiator enabled trypsin to be functionalised with sites for polymer growth. MALDI-TOF MS (Table 1) indicated ∼5 initiator sites were attached to trypsin, and the AGET ATRP method enabled the smooth growth of a (PEGMA-EE-246)85:(PEGMA-ME-475)15 co-polymer. The resulting diblock, Hybrid I, was split into two batches, one of which was used to reinitiate further polymerisation with PEGMA-ME-475 to produce the triblock, Hybrid II. The conditions used for these reactions were especially important to ensure the stability of trypsin, as using lower temperature decreases the rate of autocatalysis and prevents aggregation. The choice of monomers was also critical,12 as ATRP in water by this route would not have been possible with other thermoresponsive PEGMA based copolymers which have been previously reported,10 owing to their lower solubilities in water compared to PEGMA-EE-246 and PEGMA-ME-475.
Confirmation of polymer growth, hybrid block formation, and reinitiation of polymerisation from Hybrid I to form Hybrid II was obtained by SDS-PAGE, and by GPC analysis of polymers cleaved from the conjugates by ammonium fluoride hydrolysis (Fig. 1d and e). Clear shifts in protein bands were observed via Coomassie staining of SDS-PAGE gels following polymer growth from trypsin. Subsequent staining by iodine revealed the PEG components of the broader bands of the PEGMA-trypsin conjugates. Lower molar mass fragments were not observed, indicating that under the polymerisation conditions fragmentation of the protein via autocatalytic breakdown of trypsin or through radical-mediated scission did not occur. Further evidence of the controlled polymerisation was that successful reinitiation from the chain-end of the poly(PEGMA-EE-246: PEGMA-ME-475) co-polymer in Hybrid I to form the polymer component of Hybrid II was possible. This was apparent in the GPC traces of polymers cleaved from each hybrid, with a higher molar mass of the mixed copolymer of poly(PEGMA-EE-246-stat-PEGMA-ME-475)-g-poly(PEGMA-ME-475) clearly observable (Fig. 1e).
These particular co-monomer compositions for the polymer parts of Hybrid I and II had been chosen based on our previous reports of statistical and block co-polymers prepared by AGET ATRP.12 We therefore expected the polymer components of the conjugates to exhibit thermoresponsive behaviour, but, as apparent from Fig. 2 the effects were different for each hybrid, and varied also with the solvation properties of buffer used. Under the high-ionic concentrations of PBS at pH 7.4, Hybrid I exhibited a slightly non-linear change in turbidity with temperature between ∼30–45 °C at concentrations of 3 mg mL−1, and a 3-fold increase in turbidity overall across this temperature range. By contrast Hybrid II showed no significant changes in turbidity across the same temperature range and at the same concentration. In the reduced ionic strength of the conventional trypsin proteolysis buffer (0.05M Tris, pH 8.2, and containing 20mM CaCl2) Hybrid I showed a sharp LCST at 34 °C (i.e. just below the LCST of the polymer after cleavage from trypsin as reported in Table 1) and a ∼20-fold increase in turbidity between 34–37 °C. Hybrid II behaved very differently, showing a very small reduction in turbidity across the same temperature range in Tris buffer (Fig. 2b). These variations in temperature-responsive behaviour were strongly suggestive of differences in block behaviour in the different conjugates. Hybrid I was expected to become an amphiphilic di-block co-polymer above the LCST of the attached poly(PEGMA-EE-246-stat-PEGMA-ME-475); the assembly into higher order structures above LCST was a likely result assuming appropriate packing of the poly(PEGMA) chains into a ‘core’. DLS data (ESI†) was supportive of this interpretation, as ∼ 10 nm diameter species were observed in Tris buffer at 30 °C (below polymer LCST), but strongly scattering particles of ∼300 nm diameter were predominant at 45 °C (above polymer LCST). PGSE-NMR provided supporting evidence for the collapse of the poly(PEGMA-EE-246-stat-PEGMA-ME-475) chains. The differences observed in extent of self-assembly for Hybrid I in the different ionic strength buffers was also suggestive of a mechanism in which the pPEGMA chains self-associated into a core, as marked salting in and salting out effects have been reported previously for related poly(PEGMA-EE-246-stat-PEGMA-ME-475) co-polymers.12 The peak in the SANS data for this hybrid are very clear evidence of such a core-shell structure.
For Hybrid II however, we expected the poly(PEGMA-EE-246-stat-PEGMA-ME-475) attached to trypsin to collapse above LCST, but the outermost block of poly(PEGMA-ME475) would not collapse under these conditions, and thus an essentially double-hydrophilic block co-polymer was anticipated irrespective of whether the temperature was above or below the LCST of the middle block. Again, the PGSE-NMR show that the poly(PEGMA-EE-246-stat-PEGMA-ME-475) component collapses with temperature. Although self-assembly of double-hydrophilic block co-polymers has been observed with other systems,54,55 the turbidimetry and DLS data suggested that for Hybrid II under the conditions described (PBS or Tris buffer, 20–45 °C), higher order structures were not formed, confirmed by the absence of the peak in the Kratky plot for this hybrid.
Cryo-TEM (Fig. 3) yielded further data that indicated differences in self-assembly modes of the hybrid bioconjugates. Although drying artefacts cannot be excluded from cryo-TEM, the apparent clusters of particles in micrographs of Hybrid I flash-frozen from above polymer LCST were suggestive of self-association. These particle clusters were visually different from particles of Hybrid I below LCST, and of Hybrid II below or above LCST. The size ranges observed in TEM (2–50 nm for Hybrid II, 50–400 nm for Hybrid I) were more indicative of clusters of clusters rather than individual polymer-trypsin particles, and in all cases were larger than those observed in DLS. However, the larger particles in a polydisperse sample would have been more likely to settle fast on a cryo-TEM grid, and thus absolute size ranges would not be expected to match those in DLS exactly. Nevertheless, the distinct appearance of Hybrid I structures from above LCST compared to all the other samples implied a different assembly mode for this polymer above LCST.
Due to the limitations of cryo-TEM, imaging of samples was carried out using AFM in dilute buffer at variable temperatures (Fig. 4 and Table 2). As apparent from Fig. 4a, an increase in temperature from 30 °C to 40 °C resulted in an increase in size of Hybrid I, and a subsequent decrease in size of particles as the sample was cooled back to 30 °C. By contrast, Hybrid II (Fig. 4b and Table 2) did not self-assemble into larger structures to any significant degree over the same temperature profiles. Grain size analysis (Table 2) indicated some dispersity in particle sizes across both polymer hybrids, thus we cannot rule out completely any self-assembly for Hybrid II, but as apparent from Fig. 4, there were clearly more, and much larger, particulate species for Hybrid I at 40 °C than for Hybrid II under the same conditions. The sizes of the Hybrid II particles measured by AFM at all temperature ranges, and for Hybrid I recorded at 30 °C (before and after heating), were slightly broader than the dimensions observed in DLS experiments before aggregation. This broadening can be attributed to the finite size of the apex of the AFM probe, which frequently results in the broadening of image features.56 However, in general AFM results were in good agreement with DLS, except for Hybrid I at elevated temperatures. Both techniques showed that aggregation occurred at 40 °C, but absolute values ranged from 10–58 nm in size as reported by AFM studies but 150 nm in DLS studies. This discrepancy may have been as a result of the swelling of the polymer chains of the conjugates in buffer in DLS studies and a hindered aggregation process in AFM studies due to the surface association of the conjugates with the mica surface; however, it may simply also have reflected the different sampling nature of AFM (individual measurements) and DLS (averaged scatter).
The possible changes in trypsin structure and the likely differences in enzyme accessibility due to self-assembly modes of the hybrids were probed by activity assays. We used the well-known small peptide analogue, BAPNA, and the protein casein, which itself is known to self-assemble into complex higher-order structures.
Native trypsin loses its integrity at raised temperature,57 the scope of which is also affected by the pH and ion environment.58 As can be observed from Fig. 6a, both Hybrid I and Hybrid II were more stable compared to native trypsin, as measured by their ability to hydrolyse BAPNA over time. Hybrid II was much more stable, which we attribute to the fact that it did not precipitate from solution over time, remained better hydrated8 and likely presented a more persistent steric shield against attack from adjacent trypsin hybrids in solution than would have been the case for native trypsin. However, both hybrids displayed lower affinities for the small molecule substrate and lower turnover rates. These data were not unanticipated, as extensive conjugation of polymer chains (∼ 5 attached to each trypsin for both hybrids in this case) and higher molecular weight of the polymers57 is known to reduce access of substrates more to binding sites in comparison to lower levels of conjugation and smaller polymers. Activities of the conjugates varied with temperature, with a greater change for Hybrid I compared to Hybrid II across the LCST. Intriguingly, for the higher molar mass substrate, casein, there was a sharp decrease in hydrolysis using Hybrid I around its LCST, but an increase in the activity of Hybrid II over the same range. We attribute this to the decreased accessibility of the larger, self-assembled Hybrid I systems above LCST compared to Hybrid II. Recent data suggests that casein micelles possess an internal structure composed of a bicontinuous system of water channels and, in the presence of calcium phosphate, strands formed of calcium phosphate/casein nanoclusters.40 Such a structure is known to be permeable to trypsin, and should allow acccess to the non-associated Hybrid II structures, but would exclude superstructures/assemblies of Hybrid I above LCST.
When considered together, the data from turbidimetry, DLS, TEM, AFM and enzyme activity suggest a model for the polymer-trypsin hybrids and their differing behaviours below and above the LCSTs of their attached polymer chains (Scheme 3).
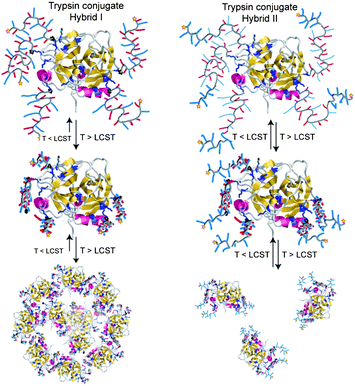 | ||
| Scheme 3 Cartoon representation of the solution behaviour of Hybrid I (left) and Hybrid II (right). | ||
We suggest that Hybrid I self-assembled and eventually aggregated above LCST due to collapse of the responsive polymer segment and the generation of exposed hydrophobic regions. It is probable that the high level of water associated with the PEGMA side chains drove the self assembly,12 where the collapse of the polymer led to high loss of water and a large change of the packing parameter. Lowering the temperature below the LCST rehydrated the polymer again allowing the protein hybrid to disassemble. It is likely then that the reduced trypsin activity of Hybrid I above the LCST arose from the aggregation and precipitation which made the enzyme less accessible than Hybrid II at the higher temperatures. By contrast, for Hybrid II the presence of the outermost block of hydrophilic PEGMA-ME-475 created a steric shield which remained chain-extended even after collapse of the intermediate block, thus disrupting hydrophobic interactions and subsequent aggregation. However, this hydrophilic outer block also rendered Hybrid II more bulky and sterically hindered access to the attached trypsin at temperatures below and above LCST. Accordingly, while Hybrid II was more stable to autocatalytic degradation it did not display temperature dependent hydrolytic activity, except for activity against casein micelles/nanoclusters where the accessibility of the substrate itself was likely to have been size-critical. The large size of the casein micelle (∼100 nm) most likely allowed access of Hybrid II, which although larger than Hybrid I below LCST, was likely to be smaller than Hybrid I above LCST as it remained unimeric, whereas the aggregation of Hybrid I into large clusters above the LCST led to a decreased accessibility of the protease towards the casein substrate. This behaviour was not observed with the small peptide substrate since the diffusion of the small peptide into the aggregate network was less likely to be as sterically hindered.
Conclusions
In conclusion, we have demonstrated the successful synthesis of “smart” PEGMA trypsin conjugates using the “growing from” approach. The polymerisations were carried out in fully aqueous solution at 4 °C using conditions mild enough to be applicable to many other sensitive proteins and biopolymers. By adopting the ATRP AGET polymerisation method to grow from the protein different polymers, two different morphologies were attainable. The morphology of the polymer was shown to affect the solution behaviour of the protein and subsequently its activity. In addition, the substrate specificity could be modified as a function of the polymer architecture. For practical applications, polymer bioconjugates need to be more active than the native protein/biopolymer, or more stable. Here we have shown that hybrid bioconjugates can be more stable, and also substrate selective, dependent on architecture, but with the penalty of reduced proteolytic activity. Accordingly, the methodology we have developed could have potential applications in the field of mass spectroscopy, drug delivery and for catalysis. In particular, these responsive hybrid conjugates could be used to target different receptors/substrates through changes in polymer architecture and external stimulus, thus enabling switchable specificity of protein activity as required.Acknowledgements
We thank the Engineering and Physical Sciences Research Council (EPSRC for grants EP/C013220/1, EP/H005625/1 and EP/G042462/1), STFC for the provision of neutron beam time, the Turkish Government (Scholarship to GY), the Australian Research Council (DP1094205, DP0880032) and the University of Nottingham for financial support. We also thank Drs George Pasparakis and Wenxin Wang for many helpful discussions, Christine Grainger-Boultby for technical support and Dr Michael Fay and Dr Christopher Parmenter for assisting with the Cryo-TEM experiments.Notes and references
- S. Frokjaer and D. E. Otzen, Nat. Rev. Drug Discovery, 2005, 4, 298–306 CrossRef CAS.
- F. M. Veronese and G. Pasut, Drug Discovery Today, 2005, 10, 1451–1458 CrossRef CAS.
- P. S. S. Allan and S. Hoffman, Macromol. Symp., 2004, 207, 139–152 CrossRef CAS.
- Z. L. Ding, G. H. Chen and A. S. Hoffman, J. Biomed. Mater. Res., 1998, 39, 498–505 CAS.
- C. Ó. F. A. Murphy, Biotechnol. Bioeng., 1998, 58, 366–373 CrossRef.
- A. Chilkoti, M. R. Dreher and D. E. Meyer, Adv. Drug Delivery Rev., 2002, 54, 1093–1111 CrossRef CAS.
- M. Heskins and J. E. Guillet, J. Macromol. Sci., Part A: Pure Appl. Chem., 1968, 2, 1441–1455 Search PubMed.
- H. Lee and T. G. Park, Biotechnol. Prog., 1998, 14, 508–516 CrossRef CAS.
- H. Vihola, A. Laukkanen, L. Valtola, H. Tenhu and J. Hirvonen, Biomaterials, 2005, 26, 3055–3064 CrossRef CAS.
- J. F. Lutz and A. Hoth, Macromolecules, 2006, 39, 893–896 CrossRef CAS.
- J.-F. Lutz, O. Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046–13047 CrossRef CAS.
- J. P. Magnusson, A. Khan, G. Pasparakis, A. O. Saeed, W. Wang and C. Alexander, J. Am. Chem. Soc., 2008, 130, 10852–10853 CrossRef CAS.
- S. R. Abulateefeh, A. O. Saeed, J. W. Aylott, W. C. Chan, M. C. Garnett, B. R. Saunders and C. Alexander, Chem. Commun., 2009, 6068–6070 RSC.
- J. P. Magnusson, S. Bersani, S. Salmaso, C. Alexander and P. Caliceti, Bioconjugate Chem., 2010, 21, 671–678 CrossRef CAS.
- Z. Zarafshani, T. Obata and J.-F. o. Lutz, Biomacromolecules, 2010, 11, 2130–2135 CrossRef CAS.
- S. M. Ryan, X. X. Wang, G. Mantovani, C. T. Sayers, D. M. Haddleton and D. J. Brayden, J. Controlled Release, 2009, 135, 51–59 CrossRef CAS.
- W. Gao, W. Liu, J. A. Mackay, M. R. Zalutsky, E. J. Toone and A. Chilkoti, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15231–15236 CrossRef CAS.
- J.-F. Lutz, Adv. Mater., 2011, 23, 2237 CrossRef CAS.
- K. L. Heredia, D. Bontempo, T. Ly, J. T. Byers, S. Halstenberg and H. D. Maynard, J. Am. Chem. Soc., 2005, 127, 16955–16960 CrossRef CAS.
- C. Boyer, V. Bulmus, J. Liu, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, J. Am. Chem. Soc., 2007, 129, 7145–7154 CrossRef CAS.
- P. De, M. Li, S. R. Gondi and B. S. Sumerlin, J. Am. Chem. Soc., 2008, 130, 11288–11289 CrossRef CAS.
- J. V. Olsen, S.-E. Ong and M. Mann, Mol. Cell. Proteomics, 2004, 3, 608–614 CrossRef CAS.
- M. A. Gauthier and H. A. Klok, Polym. Chem., 2010, 1, 1352–1373 RSC.
- A. Mero, G. Pasut, L. D. Via, M. W. M. Fijten, U. S. Schubert, R. Hoogenboom and F. M. Veronese, J. Controlled Release, 2008, 125, 87–95 CrossRef CAS.
- M. Yan, J. Ge, W. G. Dong, Z. Liu and P. Ouyang, Biochem. Eng. J., 2006, 30, 48–54 CrossRef CAS.
- K. L. Heredia, D. Bontempo, T. Ly, J. T. Byers, S. Halstenberg and H. D. Maynard, J. Am. Chem. Soc., 2005, 127, 16955–16960 CrossRef CAS.
- Y. Hao, M. Andersson, C. Virto, I. Y. Galaev, B. Mattiasson and R. Hatti-Kaul, Biocatal. Biotransform., 2001, 19, 341–359 CrossRef CAS.
- Z. Tyeklar, R. R. Jacobson, N. Wei, N. N. Murthy, J. Zubieta and K. D. Karlin, J. Am. Chem. Soc., 1993, 115, 2677–2689 CrossRef CAS.
- U. Jacquemard, V. Bénéteau, M. Lefoix, S. Routier, J.-Y. Mérour and G. Coudert, Tetrahedron, 2004, 60, 10039–10047 CrossRef CAS.
- F. M. B. Ausubel, R. Kingston, R. E. Moore, D. D. Seidman, J. G. Smith, J. A. Struhl, and K, Short Protocols in Molecular Biology, 5th edn, Wiley, 2002 Search PubMed.
- M. M. Kurfürst, Anal. Biochem., 1992, 200, 244–248 CrossRef.
- B. S. Lele, H. Murata, K. Matyjaszewski and A. J. Russell, Biomacromolecules, 2005, 6, 3380–3387 CrossRef CAS.
- J. A. Davies and P. C. Griffiths, Macromolecules, 2003, 36, 950–952 CrossRef CAS.
- P. K. Smith, R. I. Krohn, G. T. Hermanson, A. K. Mallia, F. H. Gartner, M. D. Provenzano, E. K. Fujimoto, N. M. Goeke, B. J. Olson and D. C. Klenk, Anal. Biochem., 1985, 150, 76–85 CrossRef CAS.
- D. Cunliffe, C. de las Heras Alarcon, V. Peters, J. R. Smith and C. Alexander, Langmuir, 2003, 19, 2888–2899 CrossRef CAS.
- B. F. Erlanger, N. Kokowsky and W. Cohen, Arch. Biochem. Biophys., 1961, 95, 271–278 CrossRef CAS.
- M. Singh and A. D. Krikorian, J. Agric. Food Chem., 1982, 30, 799–800 CrossRef CAS.
- S. Shaunak, A. Godwin, J. W. Choi, S. Balan, E. Pedone, D. Vijayarangam, S. Heidelberger, I. Teo, M. Zloh and S. Brocchini, Nat. Chem. Biol., 2006, 2, 312–313 CrossRef CAS.
- V. Arasaratnam, I. Y. Galaev and B. Mattiasson, Enzyme Microb. Technol., 2000, 27, 254–263 CrossRef CAS.
- D. G. Dalgleish, Soft Matter, 2011, 7, 2265–2272 RSC.
- H. M. Li, A. P. Bapat, M. Li and B. S. Sumerlin, Polym. Chem., 2011, 2, 323–327 RSC.
- M. Li, H. M. Li, P. De and B. S. Sumerlin, Macromol. Rapid Commun., 2011, 32, 354–359 CrossRef CAS.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Prog. Polym. Sci., 2010, 35, 278–301 CrossRef CAS.
- L. A. Canalle, D. Lowik and J. C. M. van Hest, Chem. Soc. Rev., 2010, 39, 329–353 RSC.
- D. Lowik, E. H. P. Leunissen, M. van den Heuvel, M. B. Hansen and J. C. M. van Hest, Chem. Soc. Rev., 2010, 39, 3394–3412 RSC.
- The terms “di-block” and “tri-block” are in quotation marks because on average 5 polymer chains were attached to each trypsin molecule, however, as the properties of each polymer chain grown from the trypsin anchor were expected to be similar, it is more convenient to refer to “di-block” than “oligo-di-block”.
- A. S. Hoffman, Clin. Chem., 2000, 46, 1478–1486 CAS.
- B. Treetharnmathurot, L. Dieudonne, E. L. Ferguson, D. Schmaljohann, R. Duncan and R. Wiwattanapatapee, Int. J. Pharm., 2009, 373, 68–76 CrossRef CAS.
- M. Matsukata, T. Aoki, K. Sanui, N. Ogata, A. Kikuchi, Y. Sakurai and T. Okano, Bioconjugate Chem., 1996, 7, 96–101 CrossRef CAS.
- S. Balan, J. W. Choi, A. Godwin, I. Teo, C. M. Laborde, S. Heidelberger, M. Zloh, S. Shaunak and S. Brocchini, Bioconjugate Chem., 2007, 18, 61–76 CrossRef CAS.
- K. L. Christman, R. M. Broyer, Z. P. Tolstyka and H. D. Maynard, J. Mater. Chem., 2007, 17, 2021–2027 RSC.
- G. H. Chen and A. S. Hoffman, Bioconjugate Chem., 1993, 4, 509–514 CrossRef CAS.
- A. S. Hoffman and P. S. Stayton, Prog. Polym. Sci., 2007, 32, 922–932 CrossRef CAS.
- G. Pasparakis and C. Alexander, Angew. Chem., Int. Ed., 2008, 47, 4847–4850 CrossRef CAS.
- W. Agut, A. Brulet, D. Taton and S. Lecommandoux, Langmuir, 2007, 23, 11526–11533 CrossRef CAS.
- D. Ricci and P. C. Braga, in Atomic Force Microscopy: Biomedical Methods and Applications, ed. D. Ricci and P. C. Braga, Humana Press Inc., Totowa, NJ, 2004, pp. 25–37 Search PubMed.
- B. Treethammathurot, C. Ovartlarnporn, J. Wungsintaweekul, R. Duncan and R. Wiwattanapatapee, Int. J. Pharm., 2008, 357, 252–259 CrossRef CAS.
- M. L. Simon, K. Laszlo, M. Kotorman and B. Szajani, Acta Biol. (Szeged), 2001, 45, 43–49 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1py00128k |
| This journal is © The Royal Society of Chemistry 2011 |